


 Sign Out
Sign Out

Adverse Reactions in Adults: The data described herein reflect exposure to REMICADE in 4779 adult patients (1304 patients with RA, 1106 patients with CD, 202 with AS, 293 with PsA, 484 with UC, 1373 with Ps, and 17 patients with other conditions), including 2625 patients exposed beyond 30 weeks and 374 exposed beyond 1 year. [For information on adverse reactions in pediatric patients, see Clinical Trials Experience: Adverse Reactions in Pediatric Patients as follows]. One of the most-common reasons for discontinuation of treatment was infusion-related reactions (e.g., dyspnea, flushing, headache and rash).
Infusion-related reactions: An infusion-related reaction was defined in clinical studies as any adverse event occurring during an infusion or within 1 hour after an infusion. In Phase III clinical studies, 18% of infliximab-treated patients compared with 5% of placebo-treated patients experienced an infusion-related reaction. Overall, a higher proportion of patients receiving infliximab monotherapy experienced an infusion-related reaction compared to patients receiving infliximab with concomitant immunomodulators. Approximately 3% of patients discontinued treatment due to infusion-related reactions and all patients recovered with or without medical therapy. Of infliximab-treated patients who had an infusion reaction during the induction period, through week 6, 27% experienced an infusion reaction during the maintenance period, week 7 through week 54. Of patients who did not have an infusion reaction during the induction period, 9% experienced an infusion reaction during the maintenance period.
In a clinical study of patients with rheumatoid arthritis (ASPIRE), infusions were to be administered over 2 hours for the first 3 infusions. The duration of subsequent infusions could be shortened to not less than 40 minutes in patients who did not experience serious infusion reactions. In this trial, sixty six percent of the patients (686 out of 1,040) received at least one shortened infusion of 90 minutes or less and 44% of the patients (454 out of 1,040) received at least one shortened infusion of 60 minutes or less. Of the infliximab-treated patients who received at least one shortened infusion, infusion-related reactions occurred in 15% of patients and serious infusion reactions occurred in 0.4% of patients.
In a clinical study of patients with Crohn's disease (SONIC), infusion-related reactions occurred in 16.6% (27/163) of patients receiving infliximab monotherapy, 5% (9/179) of patients receiving infliximab in combination with AZA, and 5.6% (9/161) of patients receiving AZA monotherapy. One serious infusion reaction (< 1%) occurred in a patient on infliximab monotherapy.
In post-marketing experience, cases of anaphylactic-like reactions, including laryngeal/pharyngeal oedema and severe bronchospasm, and seizure have been associated with Remicade administration (see Postmarketing Experience as follows).
Cases of transient visual loss occurring during or within 2 hours of Remicade infusion have been reported. Events (some fatal) of myocardial ischaemia/infarction and arrhythmia have been reported, some in close temporal association with infusion of infliximab; cerebrovascular accidents have also been reported in close temporal association with infusion of infliximab.
Among all REMICADE infusions, 3% were accompanied by nonspecific symptoms such as fever or chills, 1% were accompanied by cardiopulmonary reactions (primarily chest pain, hypotension, hypertension or dyspnea), and <1% were accompanied by pruritus, urticaria, or the combined symptoms of pruritus/urticaria and cardiopulmonary reactions. Serious infusion reactions occurred in <1% of patients and included anaphylaxis, convulsions, erythematous rash and hypotension. Approximately 3% of patients discontinued REMICADE because of infusion reactions, and all patients recovered with treatment and/or discontinuation of the infusion. REMICADE infusions beyond the initial infusion were not associated with a higher incidence of reactions. The infusion reaction rates remained stable in Ps through 1 year in Ps Study I. In psoriasis Study II, the rates were variable over time and somewhat higher following the final infusion than after the initial infusion. Across the 3 Ps studies, the percent of total infusions resulting in infusion reactions (i.e., an adverse event occurring within 1 hour) was 7% in the 3 mg/kg group, 4% in the 5 mg/kg group, and 1% in the placebo group.
Patients who became positive for antibodies to infliximab were more likely (approximately two-to three-fold) to have an infusion reaction than were those who were negative. Use of concomitant immunosuppressant agents appeared to reduce the frequency of both antibodies to infliximab and infusion reactions [see Immunogenicity as follows; Immunosuppressants under Interactions].
Infusion Reactions Following Re-administration: In a clinical trial of patients with moderate to severe Ps designed to assess the efficacy of long-term maintenance therapy versus re-treatment with an induction regimen of REMICADE following disease flare, 4% (8/219) of patients in the re-treatment induction therapy arm experienced serious infusion reactions versus <1% (1/222) in the maintenance therapy arm. Patients enrolled in this trial did not receive any concomitant immunosuppressant therapy. In this study, the majority of serious infusion reactions occurred during the second infusion at Week 2. Symptoms included, but were not limited to, dyspnea, urticaria, facial edema, and hypotension. In all cases, REMICADE treatment was discontinued and/or other treatment instituted with complete resolution of signs and symptoms.
Delayed Reactions/Reactions Following Re-administration: In Ps studies, approximately 1% of REMICADE-treated patients experienced a possible delayed hypersensitivity reaction, generally reported as serum sickness or a combination of arthralgia and/or myalgia with fever and/or rash. These reactions generally occurred within 2 weeks after repeat infusion.
Infections: In REMICADE clinical studies, treated infections were reported in 36% of REMICADE-treated patients (average of 51 weeks of follow-up) and in 25% of placebo-treated patients (average of 37 weeks of follow-up). The infections most frequently reported were respiratory tract infections (including sinusitis, pharyngitis, and bronchitis) and urinary tract infections. Among REMICADE-treated patients, serious infections included pneumonia, cellulitis, abscess, skin ulceration, sepsis, and bacterial infection. In clinical trials, 7 opportunistic infections were reported; 2 cases each of coccidioidomycosis (1 case was fatal) and histoplasmosis (1 case was fatal), and 1 case each of pneumocystosis, nocardiosis and cytomegalovirus. Tuberculosis (TB) was reported in 14 patients, 4 of whom died due to miliary tuberculosis. Other cases of TB, including disseminated TB, also have been reported post-marketing. Most of these cases of TB occurred within the first 2 months after initiation of therapy with REMICADE and may reflect recrudescence of latent disease [see Serious Infections under Precautions]. In the 1-year placebo-controlled studies RA I and RA II, 5.3% of patients receiving REMICADE every 8 weeks with MTX developed serious infections as compared to 3.4% of placebo patients receiving MTX. Of 924 patients receiving REMICADE, 1.7% developed pneumonia and 0.4% developed TB, when compared to 0.3% and 0.0% in the placebo arm respectively. In a shorter (22-week) placebo-controlled study of 1082 RA patients randomized to receive placebo, 3 mg/kg or 10 mg/kg REMICADE infusions at 0, 2, and 6 weeks, followed by every 8 weeks with MTX, serious infections were more frequent in the 10 mg/kg REMICADE group (5.3%) than the 3 mg/kg or placebo groups (1.7% in both). During the 54-week Crohn's II Study, 15% of patients with fistulizing CD developed a new fistula-related abscess.
In REMICADE clinical studies in patients with UC, infections treated with antimicrobials were reported in 27% of REMICADE-treated patients (average of 41 weeks of follow-up) and in 18% of placebo-treated patients (average 32 weeks of follow-up). The types of infections, including serious infections, reported in patients with UC were similar to those reported in other clinical studies.
The onset of serious infections may be preceded by constitutional symptoms such as fever, chills, weight loss, and fatigue. The majority of serious infections, however, may also be preceded by signs or symptoms localized to the site of the infection.
Autoantibodies/Lupus-like Syndrome: Approximately half of REMICADE-treated patients in clinical trials who were antinuclear antibody (ANA) negative at baseline developed a positive ANA during the trial compared with approximately one-fifth of placebo-treated patients. Anti-dsDNA antibodies were newly detected in approximately one-fifth of REMICADE-treated patients compared with 0% of placebo-treated patients. Reports of lupus and lupus-like syndromes, however, remain uncommon.
Malignancies: In controlled trials, more REMICADE-treated patients developed malignancies than placebo-treated patients [see Malignancies under Precautions].
In a randomized controlled clinical trial exploring the use of REMICADE in patients with moderate to severe COPD who were either current smokers or ex-smokers, 157 patients were treated with REMICADE at doses similar to those used in RA and CD. Of these REMICADE-treated patients, 9 developed a malignancy, including 1 lymphoma, for a rate of 7.67 cases per 100 patient-years of follow-up (median duration of follow-up 0.8 years; 95% CI 3.51 -14.56). There was 1 reported malignancy among 77 control patients for a rate of 1.63 cases per 100 patient-years of follow-up (median duration of follow-up 0.8 years; 95% CI 0.04 -9.10). The majority of the malignancies developed in the lung or head and neck [see Malignancies under Precautions].
Adverse Reactions in Patients with NYHA Class III/IV Heart Failure: In a randomized, double-blind study evaluating REMICADE in moderate or severe heart failure (NYHA Class III/IV; left ventricular ejection fraction ≤ 35%), 150 patients were randomized to receive treatment with 3 infusions of REMICADE 10 mg/kg, 5 mg/kg, or placebo, at 0, 2, and 6 weeks. Higher incidences of mortality and hospitalization due to worsening heart failure were observed in patients receiving the 10 mg/kg REMICADE dose. At 1 year, 8 patients in the 10 mg/kg REMICADE group had died compared with 4 deaths each in the 5 mg/kg REMICADE and the placebo groups. There were trends toward increased dyspnea, hypotension, angina, and dizziness in both the 10 mg/kg and 5 mg/kg REMICADE treatment groups, versus placebo. REMICADE has not been studied in patients with mild heart failure (NYHA Class I/II) [see Contraindications; Heart Failure under Precautions].
Hepatotoxicity: Severe liver injury, including acute liver failure and autoimmune hepatitis, has been reported in patients receiving REMICADE [see Hepatotoxicity under Precautions]. Reactivation of hepatitis B virus has occurred in patients receiving TNF blockers, including REMICADE, who are chronic carriers of this virus [see Hepatitis B Virus Reactivation under Precautions].
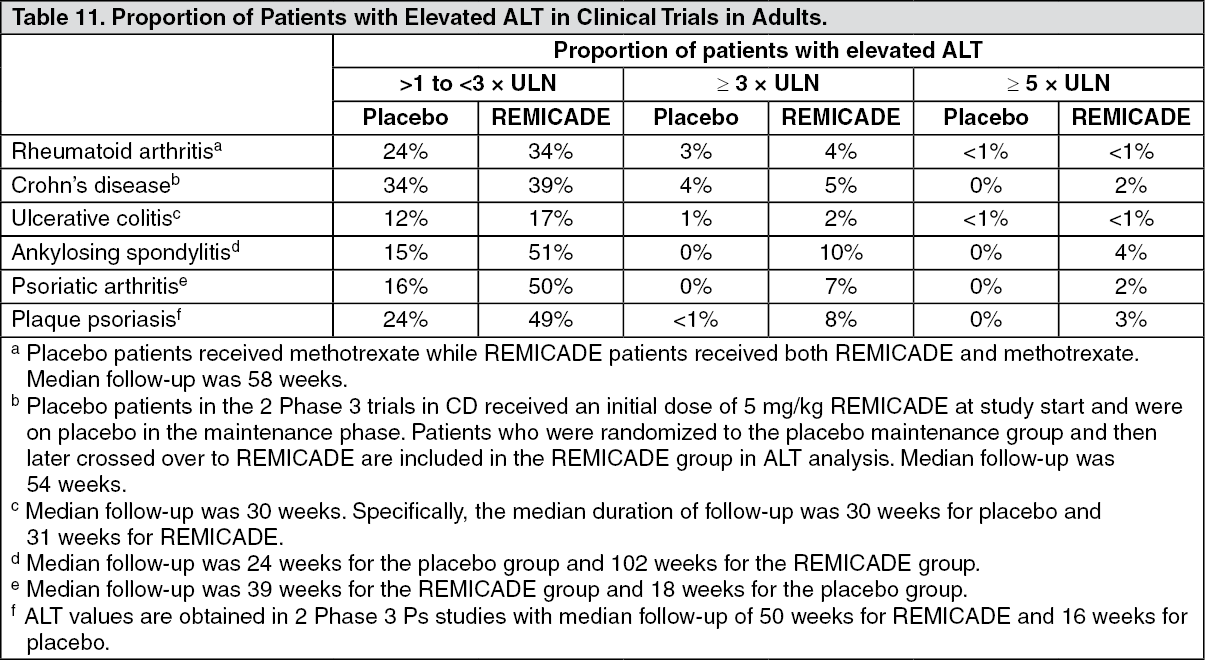
In clinical trials in RA, CD, UC, AS, Ps, and PsA, elevations of aminotransferases were observed (ALT more common than AST) in a greater proportion of patients receiving REMICADE than in controls (Table 11), both when REMICADE was given as monotherapy and when it was used in combination with other immunosuppressive agents. In general, patients who developed ALT and AST elevations were asymptomatic, and the abnormalities decreased or resolved with either continuation or discontinuation of REMICADE, or modification of concomitant medications. (See Table 11.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdverse Reactions in Psoriasis Studies: During the placebo-controlled portion across the 3 clinical trials up to Week 16, the proportion of patients who experienced at least 1 serious adverse reaction (SAE; defined as resulting in death, life threatening, requires hospitalization, or persistent or significant disability/incapacity) was 0.5% in the 3 mg/kg REMICADE group, 1.9% in the placebo group, and 1.6% in the 5 mg/kg REMICADE group.
Among patients in the 2 Phase 3 studies, 12.4% of patients receiving REMICADE 5 mg/kg every 8 weeks through 1 year of maintenance treatment experienced at least 1 SAE in Study I. In Study II, 4.1% and 4.7% of patients receiving REMICADE 3 mg/kg and 5 mg/kg every 8 weeks, respectively, through 1 year of maintenance treatment experienced at least 1 SAE.
One death due to bacterial sepsis occurred 25 days after the second infusion of 5 mg/kg REMICADE. Serious infections included sepsis, and abscesses. In Study I, 2.7% of patients receiving REMICADE 5 mg/kg every 8 weeks through 1 year of maintenance treatment experienced at least 1 serious infection. In Study II, 1.0% and 1.3% of patients receiving REMICADE 3 mg/kg and 5 mg/kg, respectively, through 1 year of treatment experienced at least 1 serious infection. The most common serious infection (requiring hospitalization) was abscess (skin, throat, and peri-rectal) reported by 5 (0.7%) patients in the 5 mg/kg REMICADE group. Two active cases of tuberculosis were reported: 6 weeks and 34 weeks after starting REMICADE.
In the placebo-controlled portion of the Ps studies, 7 of 1123 patients who received REMICADE at any dose were diagnosed with at least one NMSC compared to 0 of 334 patients who received placebo.
In the Ps studies, 1% (15/1373) of patients experienced serum sickness or a combination of arthralgia and/or myalgia with fever, and/or rash, usually early in the treatment course. Of these patients, 6 required hospitalization due to fever, severe myalgia, arthralgia, swollen joints, and immobility.
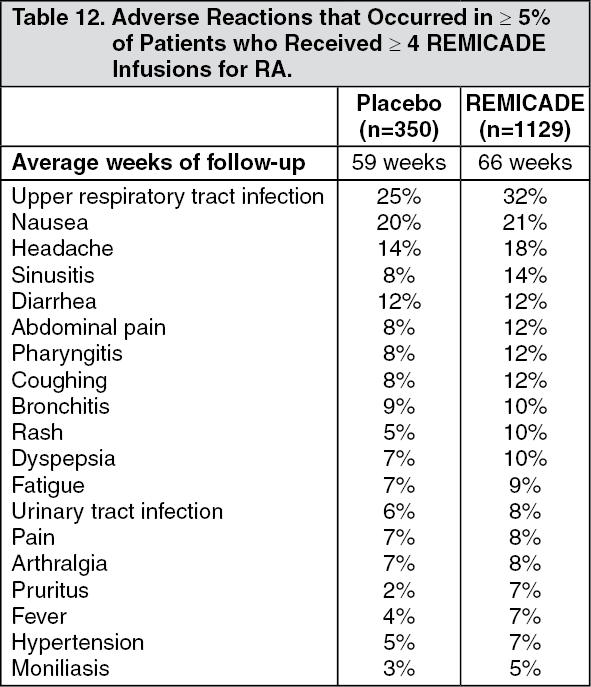
Other Adverse Reactions in Adults: Safety data are available from 4779 REMICADE-treated adult patients, including 1304 with RA, 1106 with CD, 484 with UC, 202 with AS, 293 with PsA, 1373 with Ps and 17 with other conditions. [For information on other adverse reactions in pediatric patients, see Clinical Trials Experience: Adverse Reactions in Pediatric Patients as follows]. Adverse reactions reported in ≥ 5% of all patients with RA receiving 4 or more infusions are in Table 12. The types and frequencies of adverse reactions observed were similar in REMICADE-treated RA, AS, PsA, Ps, and CD patients except for abdominal pain, which occurred in 26% of REMICADE-treated patients with CD. In the CD studies, there were insufficient numbers and duration of follow-up for patients who never received REMICADE to provide meaningful comparisons. (See Table 12.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe most common serious adverse reactions observed in clinical trials were infections [see Clinical Trials Experience: Infections as previously mentioned]. Other serious, medically relevant adverse reactions ≥ 0.2% or clinically significant adverse reactions by body system were as follows: Body as a whole: allergic reaction, edema.
Blood: pancytopenia.
Cardiovascular: hypotension.
Gastrointestinal: constipation, intestinal obstruction.
Central and Peripheral Nervous: dizziness.
Heart Rate and Rhythm: bradycardia.
Liver and Biliary: hepatitis.
Metabolic and Nutritional: dehydration.
Platelet, Bleeding and Clotting: thrombocytopenia.
Neoplasms: lymphoma.
Red Blood Cell: anemia, hemolytic anemia.
Resistance Mechanism: cellulitis, sepsis, serum sickness, sarcoidosis.
Respiratory: lower respiratory tract infection (including pneumonia), pleurisy, pulmonary edema.
Skin and Appendages: increased sweating.
Vascular (Extracardiac): thrombophlebitis.
White Cell and Reticuloendothelial: leukopenia, lymphadenopathy.
Adverse Reactions in Pediatric Patients: Adverse Reactions in Pediatric Patients with Crohn's Disease: There were some differences in the adverse reactions observed in the pediatric patients receiving REMICADE compared to those observed in adults with CD. These differences are discussed in the following paragraphs.
The following adverse reactions were reported more commonly in 103 randomized pediatric CD patients administered 5 mg/kg REMICADE through 54 weeks than in 385 adult CD patients receiving a similar treatment regimen: anemia (11%), leukopenia (9%), flushing (9%), viral infection (8%), neutropenia (7%), bone fracture (7%), bacterial infection (6%), and respiratory tract allergic reaction (6%).
Infections were reported in 56% of randomized pediatric patients in Study Peds Crohn's and in 50% of adult patients in Study Crohn's I. In Study Peds Crohn's, infections were reported more frequently for patients who received every 8-week as opposed to every 12-week infusions (74% and 38%, respectively), while serious infections were reported for 3 patients in the every 8-week and 4 patients in the every 12-week maintenance treatment group. The most commonly reported infections were upper respiratory tract infection and pharyngitis, and the most commonly reported serious infection was abscess. Pneumonia was reported for 3 patients, (2 in the every 8-week and 1 in the every 12-week maintenance treatment groups). Herpes zoster was reported for 2 patients in the every 8-week maintenance treatment group.
In Study Peds Crohn's, 18% of randomized patients experienced 1 or more infusion reactions, with no notable difference between treatment groups. Of the 112 patients in Study Peds Crohn's, there were no serious infusion reactions, and 2 patients had non-serious anaphylactoid reactions.
Elevations of ALT up to 3 times the upper limit of normal (ULN) were seen in 18% of pediatric patients in CD clinical trials; 4% had ALT elevations ≥ 3 × ULN, and 1% had elevations ≥ 5 × ULN. (Median follow-up was 53 weeks.)
Adverse Reactions in Pediatric Patients with Ulcerative Colitis: Overall, the adverse reactions reported in the pediatric UC trial and adult UC (Study UC I and Study UC II) studies were generally consistent. In a pediatric UC trial, the most common adverse reactions were upper respiratory tract infection, pharyngitis, abdominal pain, fever, and headache.
Infections were reported in 31 (52%) of 60 treated patients in the pediatric UC trial and 22 (37%) required oral or parenteral antimicrobial treatment. The proportion of patients with infections in the pediatric UC trial was similar to that in the pediatric CD study (Study Peds Crohn's) but higher than the proportion in the adults' UC studies (Study UC I and Study UC II). The overall incidence of infections in the pediatric UC trial was 13/22 (59%) in the every 8 week maintenance treatment group. Upper respiratory tract infection (7/60 [12%]) and pharyngitis (5/60 [8%]) were the most frequently reported respiratory system infections. Serious infections were reported in 12% (7/60) of all treated patients.
Elevations of ALT up to 3 times the upper limit of normal (ULN) were seen in 17% (10/60) of pediatric patients in the pediatric UC trial; 7% (4/60) had ALT elevations ≥ 3 × ULN, and 2% (1/60) had elevations ≥ 5 × ULN (median follow-up was 49 weeks).
Overall, 8 of 60 (13%) treated patients experienced one or more infusion reactions, including 4 of 22 (18%) patients in the every 8-week treatment maintenance group. No serious infusion reactions were reported.
In the pediatric UC trial, 45 patients were in the 12 to 17 year age group and 15 in the 6 to 11 year age group. The numbers of patients in each subgroup are too small to make any definitive conclusions about the effect of age on safety events. There were higher proportions of patients with serious adverse events (40% vs. 18%) and discontinuation due to adverse events (40% vs. 16%) in the younger age group than in the older age group. While the proportion of patients with infections was also higher in the younger age group (60% vs. 49%), for serious infections, the proportions were similar in the two age groups (13% in the 6 to 11 year age group vs. 11% in the 12 to 17 year age group). Overall proportions of adverse reactions, including infusion reactions, were similar between the 6 to 11 and 12 to 17 year age groups (13%).
Immunogenicity: As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described as follows with the incidence of antibodies in other studies or to other infliximab products may be misleading.
Treatment with REMICADE can be associated with the development of antibodies to infliximab. An enzyme immunoassay (EIA) method was originally used to measure anti-infliximab antibodies in clinical studies of REMICADE. The EIA method is subject to interference by serum infliximab, possibly resulting in an underestimation of the rate of patient antibody formation. A separate, drug-tolerant electrochemiluminescence immunoassay (ECLIA) method for detecting antibodies to infliximab was subsequently developed and validated. This method is 60-fold more sensitive than the original EIA. With the ECLIA method, all clinical samples can be classified as either positive or negative for antibodies to infliximab without the need for the inconclusive category.
The incidence of antibodies to infliximab was based on the original EIA method in all clinical studies of REMICADE except for the Phase 3 study in pediatric patients with UC where the incidence of antibodies to infliximab was detected using both the EIA and ECLIA methods.
Immunogenicity in Adult Patients: The incidence of antibodies to infliximab in patients with RA and CD given a 3-dose induction regimen followed by maintenance dosing was approximately 10% as assessed through 1 to 2 years of REMICADE treatment. A higher incidence of antibodies to infliximab was observed in CD patients receiving REMICADE after drug-free intervals >16 weeks. In a PsA study in which 191 patients received 5 mg/kg with or without MTX, antibodies to infliximab occurred in 15% of patients. The majority of antibody-positive patients had low titers. Antibody development was lower among RA and CD patients receiving immunosuppressant therapies such as 6-MP/AZA or MTX. Patients who were antibody-positive were more likely to have higher rates of clearance, have reduced efficacy, and to experience an infusion reaction than were patients who were antibody negative [see Clinical Trials Experience: Infusion-related reactions as previously mentioned]. In the Ps Study II, which included both the 5 mg/kg and 3 mg/kg doses, antibodies were observed in 36% of patients treated with 5 mg/kg every 8 weeks for 1 year, and in 51% of patients treated with 3 mg/kg every 8 weeks for 1 year.
In the Ps Study III, which also included both the 5 mg/kg and 3 mg/kg doses, antibodies were observed in 20% of patients treated with 5 mg/kg induction (weeks 0, 2 and 6), and in 27% of patients treated with 3 mg/kg induction. Despite the increase in antibody formation, the infusion reaction rates in Studies I and II in patients treated with 5 mg/kg induction followed by every 8 week maintenance for 1 year and in Study III in patients treated with 5 mg/kg induction (14.1%-23.0%) and serious infusion reaction rates (<1%) were similar to those observed in other study populations. The clinical significance of apparent increased immunogenicity on efficacy and infusion reactions in Ps patients as compared to patients with other diseases treated with REMICADE over the long term is not known.
Immunogenicity in Pediatric Patients with Crohn's Disease: In Study Peds Crohn's, in which all patients received stable doses of 6-MP, AZA, or MTX, excluding inconclusive samples, 3 of 24 patients had antibodies to infliximab. Although 105 patients were tested for antibodies to infliximab, 81 patients were classified as inconclusive because they could not be ruled as negative due to assay interference by the presence of infliximab in the sample.
Immunogenicity in Pediatric Patients with Ulcerative Colitis: In the pediatric UC trial, 58 patients were evaluated for antibodies to infliximab using the EIA as well as the drug-tolerant ECLIA. With the EIA, 4 of 58 (7%) patients had antibodies to infliximab. With the ECLIA, 30 of 58 (52%) patients had antibodies to infliximab. The higher incidence of antibodies to infliximab by the ECLIA method was due to the 60-fold higher sensitivity compared to the EIA method. While EIA-positive patients generally had undetectable trough infliximab concentrations, ECLIA-positive patients could have detectable trough concentrations of infliximab because the ECLIA assay is more sensitive and drug-tolerant.
Postmarketing Experience: Adverse reactions, some with fatal outcomes, have been identified during post approval use of REMICADE in adult and pediatric patients. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Postmarketing Adverse Reactions in Adults and Pediatric Patients: Neutropenia [see Hematologic Reactions under Precautions], agranulocytosis (including infants exposed in utero to infliximab), idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura.
Interstitial lung disease (including pulmonary fibrosis/interstitial pneumonitis and rapidly progressive disease).
Pericardial effusion, systemic and cutaneous vasculitis.
Erythema multiforme, Stevens-Johnson Syndrome, toxic epidermal necrolysis, linear IgA bullous dermatosis (LABD), acute generalized exanthematous pustulosis (AGEP), new onset and worsening psoriasis (all subtypes including pustular, primarily palmoplantar), lichenoid reactions.
Peripheral demyelinating disorders (such as Guillain-Barré syndrome, chronic inflammatory demyelinating polyneuropathy, and multifocal motor neuropathy) transverse myelitis, and neuropathies (additional neurologic reactions have also been observed) [see Neurologic Reactions under Precautions].
Acute liver failure, jaundice, hepatitis, and cholestasis [see Hepatotoxicity under Precautions].
Serious infections [see Serious Infections under Precautions] and vaccine breakthrough infection including bovine tuberculosis (disseminated BCG infection) following vaccination in an infant exposed in utero to infliximab [see Vaccinations and Use of Live Vaccines/Therapeutic Infectious Agents under Precautions].
Malignancies, including leukemia, melanoma, Merkel cell carcinoma, and cervical cancer [see Malignancies under Precautions].
Anaphylactic reactions, including anaphylactic shock, laryngeal/pharyngeal edema and severe bronchospasm, and seizure have been associated with REMICADE administration.
Transient visual loss have been reported in association with REMICADE during or within 2 hours of infusion. Cerebrovascular accidents, myocardial ischemia/infarction (some fatal), and arrhythmia occurring within 24 hours of initiation of infusion have also been reported [see Cardiovascular and Cerebrovascular Reactions During and After Infusion under Precautions].
Postmarketing Serious Adverse Reactions in Pediatric Patients: The following serious adverse reactions have been reported in the post-marketing experience in pediatric patients: infections (some fatal) including opportunistic infections and tuberculosis, infusion reactions, hypersensitivity reactions, malignancies, including hepatosplenic T-cell lymphomas [see Warnings; Malignancies under Precautions], transient hepatic enzyme abnormalities, lupus-like syndromes, and the development of autoantibodies.
View ADR Monitoring Form