Pharmacotherapeutic group: antineoplastic agents; other antineoplastic agents; monoclonal antibodies.
ATC code: L01FX14.
Pharmacology: Pharmacodynamics: Mechanism of action: Polatuzumab vedotin is a CD79b-targeted antibody-drug conjugate that preferentially delivers a potent anti-mitotic agent (monomethyl auristatin E, or MMAE) to B-cells, which results in the killing of malignant B-cells. The polatuzumab vedotin molecule consists of MMAE covalently attached to a humanized immunoglobulin G1 monoclonal antibody via a cleavable linker. The monoclonal antibody binds with high affinity and selectivity to CD79b, a cell surface component of the B-cell receptor. CD79b expression is restricted to normal cells within the B-cell lineage (with the exception of plasma cells) and malignant B-cells; it is expressed in >95% of diffuse large B-cell lymphoma. Upon binding CD79b, polatuzumab vedotin is rapidly internalized and the linker is cleaved by lysosomal proteases to enable intracellular delivery of MMAE. MMAE binds to microtubules and kills dividing cells by inhibiting cell division and inducing apoptosis.
Pharmacodynamic effects: Cardiac electrophysiology: Polatuzumab vedotin did not prolong the mean QTc interval to any clinically relevant extent based on ECG data from two open-label studies in patients with previously treated B-cell malignancies at the recommended dosage.
Clinical efficacy and safety: Previously untreated DLBCL: The efficacy of Polivy was evaluated in an international, multicenter, randomized double-blind, placebo-controlled study (POLARIX, GO39942) in 879 patients with previously untreated DLBCL.
Eligible patients were age 18-80, and had IPI score 2-5, and ECOG Performance Status 0-2. Histologies included DLBCL (not otherwise specified (NOS), activated B-cell (ABC), germinal center B-cell (GCB)), HGBL (NOS, double-hit, triple-hit), and other large B-cell lymphoma subtypes (EBV positive, T-cell rich/histiocyte rich). Patients did not have known CNS lymphoma or peripheral neuropathy > Grade 1.
Patients were randomized 1:1 to receive Polivy plus R-CHP or R-CHOP for six 21-day cycles followed by two additional cycles of rituximab alone in both arms. Patients were stratified by IPI score (2 vs 3-5), presence or absence of bulky disease (lesion ≥ 7.5 cm), and geographical region.
Polivy was administered intravenously at 1.8 mg/kg on Day 1 of Cycles 1-6. R-CHP or R-CHOP were administered starting on Day 1 of Cycles 1-6 followed by rituximab alone on Day 1 of Cycles 7-8.
Dosing in each treatment arm was administered according to the following: Polivy + R-CHP arm: Polivy 1.8 mg/kg, rituximab 375 mg/m
2, cyclophosphamide 750 mg/m
2, doxorubicin 50 mg/m
2, and prednisone 100 mg/day, on Days 1-5 of every cycle, orally.
R-CHOP arm: rituximab 375 mg/m
2, cyclophosphamide 750 mg/m
2, doxorubicin 50 mg/m
2, vincristine 1.4 mg/m
2, and prednisone 100 mg/day, on Days 1-5 of every cycle, orally.
The two treatment groups were generally balanced with respect to baseline demographics and disease characteristics. The median age was 65 years (range 19 to 80 years), 53.6% of patients were white and 53.8% were male, 43.8% had bulky disease, 38.0% had IPI score 2, 62.0% had IPI score 3-5, and 88.7% had Stage 3 or 4 disease. The majority of patients (84.2%) had DLBCL (including NOS, ABC, and GCB).
211 patients did not have a cell of origin (COO) result reported. Of the COO evaluable population (n=668), 33.1% of patients had ABC like DLBCL and 52.7% of patients had GCB like DLBCL, by gene expression profiling.
The primary endpoint of the study was investigator-assessed progression free survival. The median duration of follow up was 28.2 months. Efficacy results are summarized in Table 1 and in Figure. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
At the interim analysis, the key secondary endpoint of OS was immature and was not statistically different [stratified hazard ratio of 0.94 (95% CI, 0.65, 1.37); p=0.7524]. (See figure.)
Click on icon to see table/diagram/image
Relapsed or refractory DLBCL: The efficacy of Polivy was evaluated in an international, multicentre, open-label study (GO29365) which included a randomized cohort of 80 patients with previously treated DLBCL. Patients were randomized 1:1 to receive Polivy plus BR or BR alone for six 21-day cycles. Patients were stratified by duration of response to last prior treatment of ≤12 months or >12 months.
Eligible patients were not candidates for autologous haematopoietic stem cell transplant (HSCT) and had relapsed or refractory disease after receiving at least one prior systemic chemotherapy regimen. The study excluded patients with prior allogeneic HSCT, central nervous system lymphoma, transformed indolent lymphoma, grade 3b FL, significant cardiovascular or pulmonary disease, active infections, AST or alanine transaminase (ALT) >2.5 x ULN or total bilirubin ≥1.5 x ULN, creatinine >1.5 x ULN (or CrCl <40 mL/min) unless due to underlying lymphoma.
Polivy was given intravenously at 1.8 mg/kg administered on Day 2 of Cycle 1 and on Day 1 of Cycles 2-6. Bendamustine was administered at 90 mg/m
2 intravenously daily on Days 2 and 3 of Cycle 1 and on Days 1 and 2 of Cycles 2-6. Rituximab was administered at 375 mg/m
2 on Day 1 of Cycles 1-6.
Among the 80 patients who were randomized to receive Polivy plus BR (n=40) or BR alone (n=40) the majority were white (71%) and male (66%). The median age was 69 years (range: 30-86 years). Sixty-four out of 80 patients (80%) had ECOG performance status (PS) of 0-1 and 14 out of 80 patients (18%) had ECOG PS of 2. The majority of patients (98%) had DLBCL not otherwise specified (NOS). Overall, 48% of patients had activated B-cell (ABC) DLBCL and 40% had germinal center B-cell like (GCB) DLBCL. Primary reasons patients were not candidates for HSCT included age (40%), insufficient response to salvage therapy (26%) and prior transplant failure (20%). The median number of prior therapies was 2 (range: 1-7), with 29% (n=23) receiving one prior therapy, 25% (n=20) receiving 2 prior therapies, and 46% (n=37) receiving 3 or more prior therapies. All except one patient in the pola+BR arm of the randomized Phase II were naïve to bendamustine treatment. 80% of patients had refractory disease. For patients who received polatuzumab vedotin plus BR and had CD3+ lymphocyte count evaluated, the absolute CD3+ lymphocyte count was >200 cells/μL in 95%, 79% and 83% of patients analyzed at prior to therapy (n=134), end of treatment (n=72) and 6 months after end of treatment (n=18), respectively.
The primary endpoint of the study was complete response (CR) rate at end of treatment (6-8 weeks after Day 1 of Cycle 6 or last study treatment) as assessed by PET-CT by an Independent Review Committee (IRC). (See Table 2.)
Click on icon to see table/diagram/image
Overall survival (OS) was an exploratory endpoint which was not type 1 error controlled. The median OS in the Polivy plus BR arm was 12.4 months (95% CI: 9.0, NE) vs 4.7 months (95% CI: 3.7, 8.3) in the control arm. The unadjusted estimate for OS HR was 0.42. When accounting for the influence of baseline covariates the OS HR was adjusted to 0.59. Covariates included primary refractory status, number of prior lines of therapy, IPI, and prior stem cell transplant.
Investigator-assessed progression free survival (PFS) was an exploratory endpoint which was not type 1 error controlled. The median PFS in the Polivy plus BR arm was 7.6 months (95% CI: 6.0, 17.0) vs 2.0 months (95% CI: 1.5, 3.7) in the control arm. The unadjusted estimate for PFS HR was 0.34.
Immunogenicity: As with all therapeutic proteins, there is the potential for an immune response in patients treated with polatuzumab vedotin. In Studies GO39442 (POLARIX) and GO29365, 1.4% (6/427) and 5.2% (12/233) of patients tested positive for antibodies against polatuzumab vedotin, respectively, of which none were positive for neutralizing antibodies. Due to the limited number of anti-polatuzumab vedotin antibody positive patients, no conclusions can be drawn concerning a potential effect of immunogenicity on efficacy or safety.
Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of incidence of antibodies to polatuzumab vedotin with the incidence of antibodies to other products may be misleading.
Paediatric population: The European Medicines Agency has waived the obligation to submit results of studies with Polivy in all subsets of the paediatric population for the treatment of mature B-cell neoplasms (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Antibody-conjugated MMAE (acMMAE) plasma exposure increased dose-proportionally over the 0.1 to 2.4 mg/kg polatuzumab vedotin dose range. After the first 1.8 mg/kg polatuzumab vedotin dose, the acMMAE mean maximum concentration (C
max) was 803 (± 233) ng/mL and the area under the concentration-time curve from time zero to infinity (AUC
inf) was 1860 (± 966) day·ng/mL. Based on the population PK analysis, Cycle 3 acMMAE AUC increased by approximately 30% over Cycle 1 AUC, and achieved more than 90% of the Cycle 6 AUC. The terminal half-life at Cycle 6 was approximately 12 days (95% CI of 8.1 - 19.5 days) for acMMAE. Based on population PK analysis, the predicted acMMAE concentration at the end of Cycle 6 is approximately 80% of the theoretical steady-state value. Exposures of unconjugated MMAE, the cytotoxic component of polatuzumab vedotin, increased dose proportionally over the 0.1 to 2.4 mg/kg polatuzumab vedotin dose range. MMAE plasma concentrations followed formation rate limited kinetics. After the first 1.8 mg/kg polatuzumab vedotin dose, the C
max was 6.82 (± 4.73) ng/mL, the time to maximum plasma concentration is approximately 2.5 days, and the terminal half-life is approximately 4 days. Plasma exposures of unconjugated MMAE are <3% of acMMAE exposures. Based on the population PK analysis there is a decrease of plasma unconjugated MMAE exposure (AUC) after repeated every-three-week dosing.
Based on population pharmacokinetics simulations, a post-hoc analysis predicted exposure to unconjugated MMAE for patients with bodyweight over 100 kg to be increased by not more than 55%.
Absorption: Polivy is administered as an intravenous infusion. There have been no studies performed with other routes of administration.
Distribution: The population estimate of central volume of distribution for acMMAE was 3.15 L, which approximated plasma volume.
In vitro, MMAE is moderately bound (71% - 77%) to human plasma proteins. MMAE does not significantly partition into human red blood cells
in vitro; the blood to plasma ratio is 0.79 to 0.98.
In vitro data indicate that MMAE is a P-gp substrate but does not inhibit P-gp at clinically relevant concentrations.
Biotransformation: Polatuzumab vedotin is expected to undergo catabolism in patients, resulting in the production of small peptides, amino acids, unconjugated MMAE, and unconjugated MMAE related catabolites. The levels of MMAE metabolites have not been measured in human plasma.
In vitro studies indicate that MMAE is a substrate for CYP3A4/5 but does not induce major CYP enzymes. MMAE is a weak time-dependent inhibitor of CYP3A4/5 but does not competitively inhibit CYP3A4/5 at clinically relevant concentrations.
MMAE does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6.
Elimination: Based on a population PK analysis, the conjugate (acMMAE) is primarily eliminated by non-specific linear clearance pathway with a value of 0.9 L/day.
In vivo studies in rats dosed with polatuzumab vedotin (radiolabel on MMAE) demonstrate that the majority of radioactivity is excreted in faeces and the minority of radioactivity is excreted in urine.
Paediatric population: No studies have been conducted to investigate the pharmacokinetics of polatuzumab vedotin in the paediatric population (<18 years old).
Elderly: Age did not have an effect on the pharmacokinetics of acMMAE and unconjugated MMAE based on population PK analyses with patients aged 19-89 years. No significant difference was observed in the pharmacokinetics of acMMAE and unconjugated MMAE among patients <65 years of age (n=394) and patients ≥65 years of age (n=495) based on population PK analyses.
Renal impairment: In patients with mild (CrCL 60-89 mL/min, n=361) or moderate (CrCL 30-59 mL/min, n=163) renal impairment, acMMAE and unconjugated MMAE exposures are similar to patients with normal renal function (CrCL ≥90 mL/min, n=356), based on population PK analyses. There are insufficient data to assess the impact of severe renal impairment (CrCL 15-29 mL/min, n=4) on PK. No data are available in patients with end-stage renal disease and/or who are on dialysis.
Hepatic impairment: In patients with mild hepatic impairment [AST or ALT >1.0 to 2.5 x ULN or total bilirubin >1.0 to 1.5 x ULN, n=133], acMMAE exposures are similar whereas unconjugated MMAE AUC are not more than 40% higher compared to patients with normal hepatic function (n=737), based on population PK analyses.
There are insufficient data to assess the impact of moderate hepatic impairment (total bilirubin >1.5 - 3 x ULN, n=11) on PK. Limited data are available in patients with severe hepatic impairment or liver transplantation.
Toxicology: Preclinical safety data: Systemic toxicity: In both rats and cynomolgus monkeys, the predominant systemic toxicities associated with administration of MMAE and polatuzumab vedotin included reversible bone marrow toxicity and associated peripheral blood cell effects.
Genotoxicity: No dedicated mutagenicity studies have been performed with polatuzumab vedotin. MMAE was not mutagenic in the bacterial reverse mutation assay (Ames test) or the L5178Y mouse lymphoma forward mutation assay.
MMAE was genotoxic in the rat bone marrow micronucleus study probably through an aneugenic mechanism. This mechanism is consistent with the pharmacological effect of MMAE as a microtubule disrupting agent.
Carcinogenicity: No dedicated carcinogenicity studies have been performed with polatuzumab vedotin and/or MMAE.
Impairment of fertility: No dedicated fertility studies in animals have been performed with polatuzumab vedotin. However, results of the 4-week rat toxicity study indicate the potential for polatuzumab vedotin to impair male reproductive function and fertility. Testicular seminiferous tubule degeneration did not reverse following a 6-week treatment-free period and correlated with decreased testes weight and gross findings at recovery necropsy of small and/or soft testes in males given ≥2 mg/kg.
Reproductive toxicity: No dedicated teratogenicity studies in animals have been performed with polatuzumab vedotin. However, treatment of pregnant rats with MMAE at 0.2 mg/kg caused embryolethality and foetal malformations (including protruding tongue, malrotated limbs, gastroschisis, and agnathia). Systemic exposure (AUC) in rats at a dose of 0.2 mg/kg MMAE is approximately 50% of the AUC in patients who received the recommended dose of 1.8 mg/kg Polivy every 21 days.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image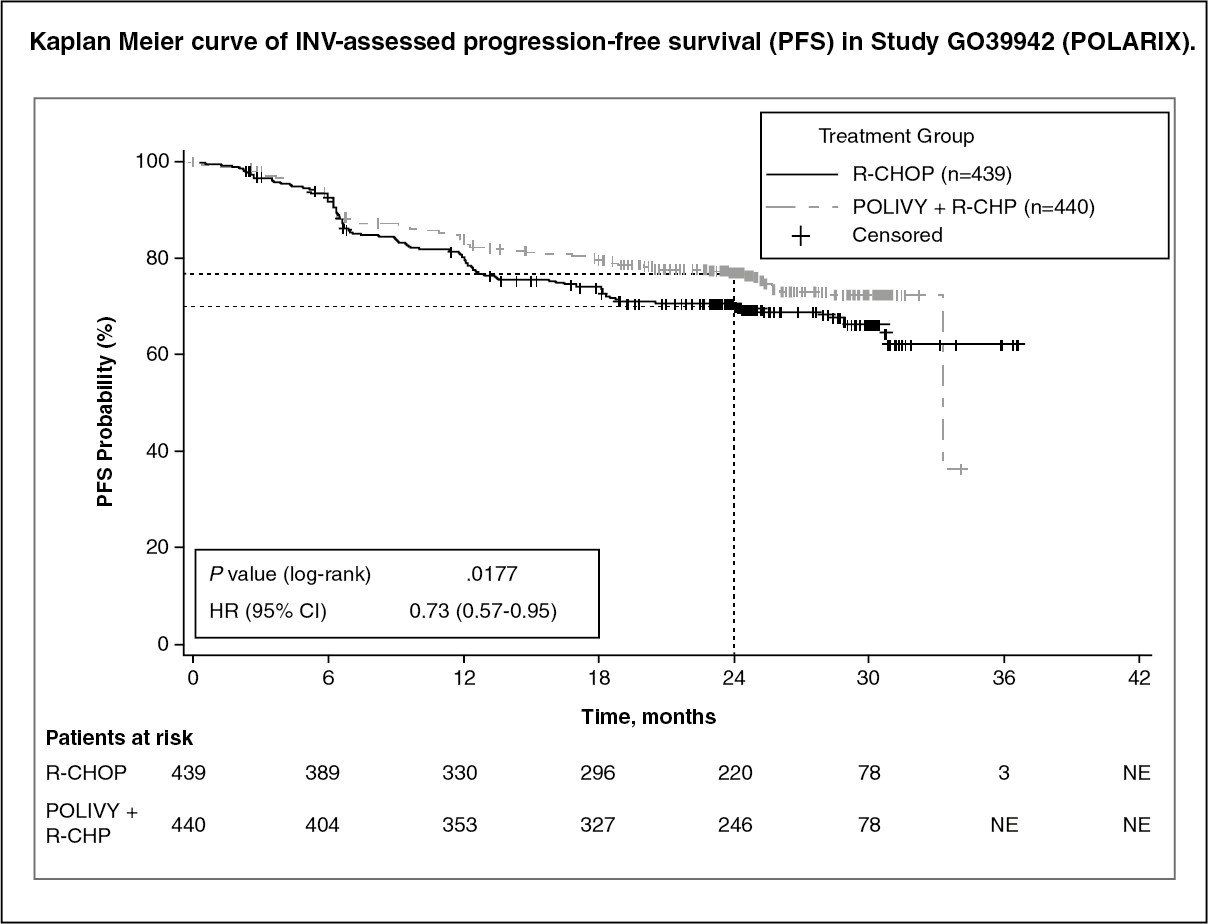 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image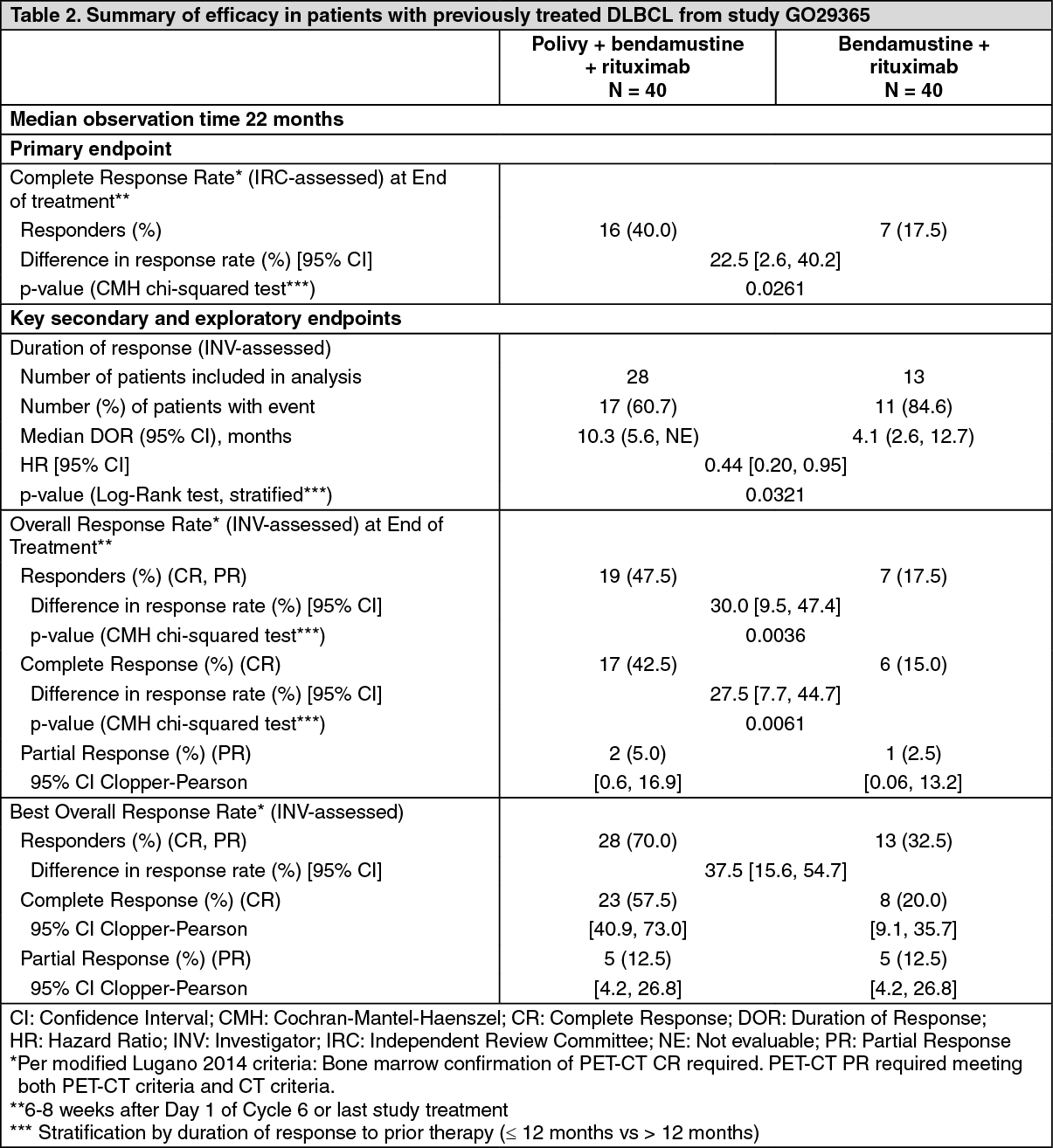 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
