


 Sign Out
Sign Out



Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in Precautions reflect exposure to LENVIMA as a single agent in 261 patients with DTC (SELECT) and 476 patients with HCC (REFLECT), LENVIMA with pembrolizumab in 406 patients with endometrial carcinoma (Study 309), LENVIMA with everolimus in 62 patients with RCC (Study 205), and LENVIMA with pembrolizumab in 352 patients with RCC (CLEAR). Safety data obtained in 1823 patients with advanced solid tumors who received LENVIMA as a single agent across multiple clinical studies was used to further characterize the risks of serious adverse reactions. Among the 1823 patients who received LENVIMA as a single agent, the median age was 61 years (20 to 89 years), the dose range was 0.2 mg to 32 mg daily, and the median duration of exposure was 5.6 months.
The following data reflect exposure to LENVIMA in 1557 patients enrolled in randomized, active-controlled trials (REFLECT; Study 205; CLEAR; Study 309), and a randomized, placebo-controlled trial (SELECT). The median duration of exposure to LENVIMA across these five studies ranged from 6 to 16 months. The demographic and exposure data for each clinical trial population are described as follows.
Differentiated Thyroid Cancer: The safety of LENVIMA was evaluated in SELECT, in which patients with radioactive iodine-refractory differentiated thyroid cancer were randomized (2:1) to LENVIMA (n=261) or placebo (n=131) [see Pharmacology: Pharmacodynamics: Clinical Studies: Differentiated Thyroid Cancer under Actions]. The median treatment duration was 16.1 months for LENVIMA. Among 261 patients who received LENVIMA, median age was 64 years, 52% were females, 80% were White, 18% were Asian, and 2% were Black; and 4% were Hispanic/Latino.
The most common adverse reactions observed in LENVIMA-treated patients (≥30%) were, in order of decreasing frequency, hypertension, fatigue, diarrhea, arthralgia/myalgia, decreased appetite, decreased weight, nausea, stomatitis, headache, vomiting, proteinuria, palmar-plantar erythrodysesthesia (PPE) syndrome, abdominal pain, and dysphonia. The most common serious adverse reactions (at least 2%) were pneumonia (4%), hypertension (3%), and dehydration (3%).
Adverse reactions led to dose reductions in 68% of patients receiving LENVIMA; 18% of patients discontinued LENVIMA for adverse reactions. The most common adverse reactions (at least 10%) resulting in dose reductions of LENVIMA were hypertension (13%), proteinuria (11%), decreased appetite (10%), and diarrhea (10%); the most common adverse reactions (at least 1%) resulting in discontinuation of LENVIMA were hypertension (1%) and asthenia (1%).
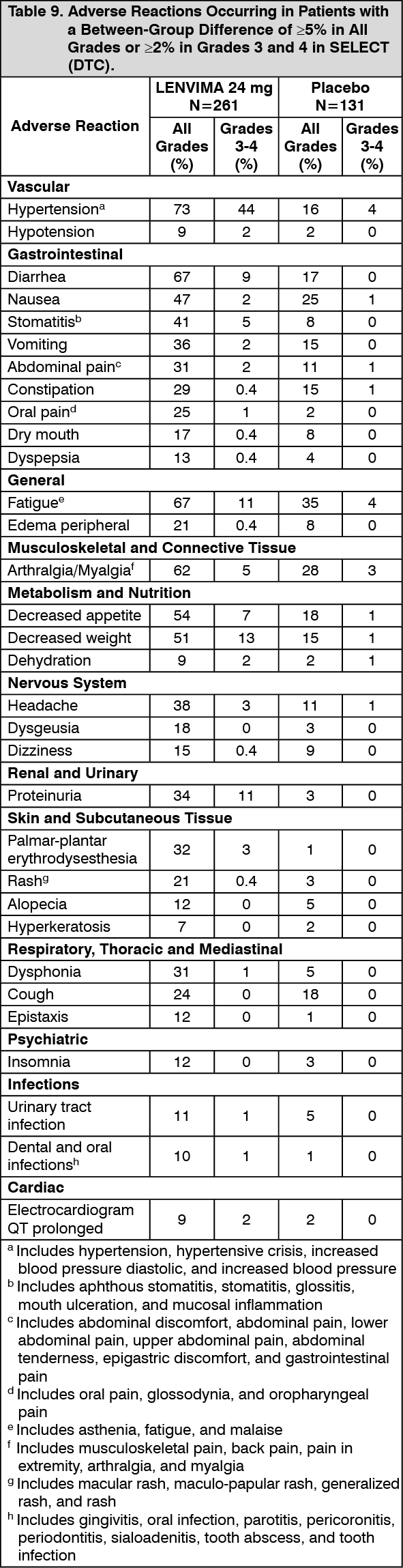
Table 9 presents adverse reactions occurring at a higher rate in LENVIMA-treated patients than patients receiving placebo in the double-blind phase of the study. (See Table 9.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageClinically important adverse reactions occurring more frequently in LENVIMA-treated patients than patients receiving placebo, but with an incidence of <5% were pulmonary embolism (3%, including fatal reports vs 2%, respectively) and osteonecrosis of the jaw (0.4% vs 0%, respectively).
Laboratory abnormalities with a difference of ≥2% in Grade 3-4 events and at a higher incidence in the LENVIMA arm are presented in Table 10. (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe following laboratory abnormalities (all Grades) occurred in >5% of LENVIMA-treated patients and at a rate that was two-fold or higher than in patients who received placebo: hypoalbuminemia, increased alkaline phosphatase, hypomagnesemia, hypoglycemia, hyperbilirubinemia, hypercalcemia, hypercholesterolemia, increased serum amylase, and hyperkalemia.
First-Line Treatment of Renal Cell Carcinoma in Combination with Pembrolizumab (CLEAR): The safety of LENVIMA in combination with pembrolizumab was investigated in CLEAR [see Pharmacology: Pharmacodynamics: Clinical Studies: Renal Cell Carcinoma under Actions]. Patients received LENVIMA 20 mg orally once daily in combination with pembrolizumab 200 mg intravenously every 3 weeks (n=352), or LENVIMA 18 mg orally once daily in combination with everolimus 5 mg orally once daily (n=355), or sunitinib 50 mg orally once daily for 4 weeks then off treatment for 2 weeks (n=340). The median duration of exposure to the combination therapy of LENVIMA and pembrolizumab was 17 months (range: 0.1 to 39).
Fatal adverse reactions occurred in 4.3% of patients receiving LENVIMA in combination with pembrolizumab, including cardio-respiratory arrest (0.9%), sepsis (0.9%), and one case (0.3%) each of arrhythmia, autoimmune hepatitis, dyspnea, hypertensive crisis, increased blood creatinine, multiple organ dysfunction syndrome, myasthenic syndrome, myocarditis, nephritis, pneumonitis, ruptured aneurysm and subarachnoid hemorrhage.
Serious adverse reactions occurred in 51% of patients receiving LENVIMA and pembrolizumab. Serious adverse reactions in ≥2% of patients were hemorrhagic events (5%), diarrhea (4%), hypertension (3%), myocardial infarction (3%), pneumonitis (3%), vomiting (3%), acute kidney injury (2%), adrenal insufficiency (2%), dyspnea (2%), and pneumonia (2%).
Permanent discontinuation of LENVIMA, pembrolizumab, or both due to an adverse reaction occurred in 37% of patients; 26% LENVIMA only, 29% pembrolizumab only, and 13% both drugs. The most common adverse reactions (≥2%) leading to permanent discontinuation of LENVIMA, pembrolizumab, or both were pneumonitis (3%), myocardial infarction (3%), hepatotoxicity (3%), acute kidney injury (3%), rash (3%), and diarrhea (2%).
Dose interruptions of LENVIMA, pembrolizumab, or both due to an adverse reaction occurred in 78% of patients receiving LENVIMA in combination with pembrolizumab. LENVIMA was interrupted in 73% of patients and both drugs were interrupted in 39% of patients. LENVIMA was dose reduced in 69% of patients. The most common adverse reactions (≥5%) resulting in dose reduction or interruption of LENVIMA were diarrhea (26%), fatigue (18%), hypertension (17%), proteinuria (13%), decreased appetite (12%), palmar-plantar erythrodysesthesia (11%), nausea (9%), stomatitis (9%), musculoskeletal pain (8%), rash (8%), increased lipase (7%), abdominal pain (6%), vomiting (6%), increased ALT (5%), and increased amylase (5%).
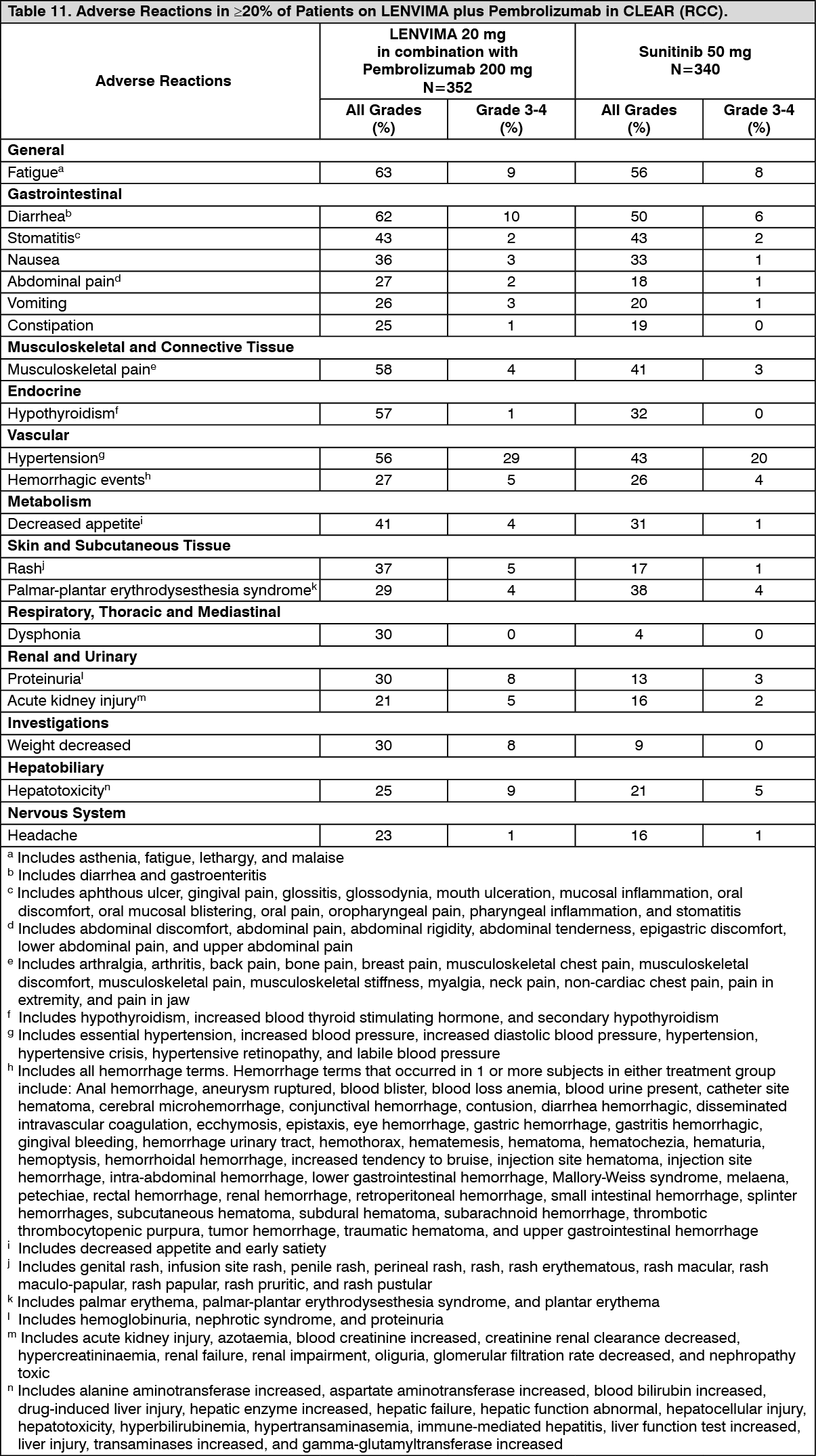
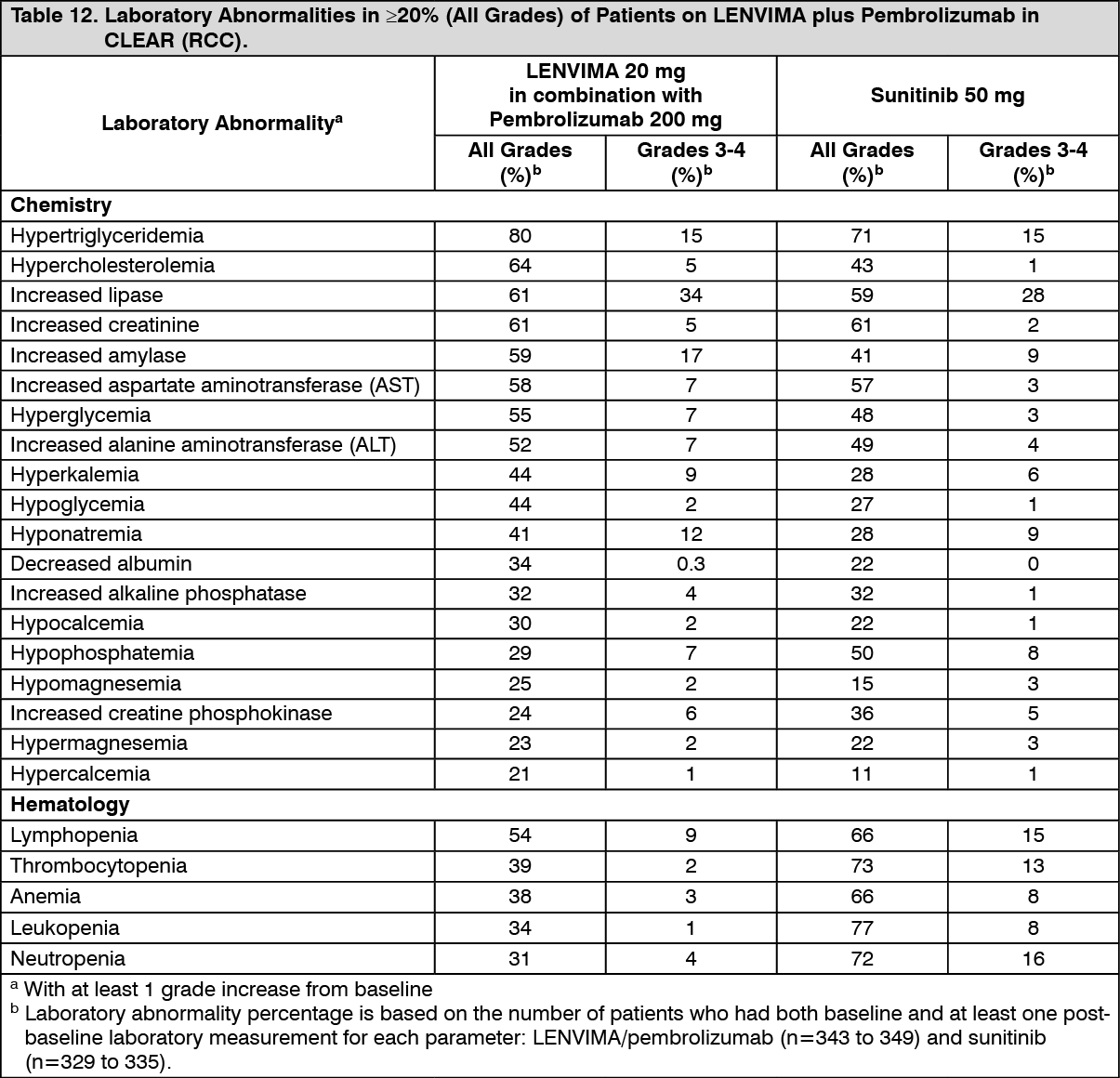
Tables 11 and 12 summarize the adverse reactions and laboratory abnormalities, respectively, that occurred in ≥20% of patients treated with LENVIMA and pembrolizumab in CLEAR. (See Tables 11 and 12.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageClinically relevant adverse reactions (<20%) that occurred in patients receiving LENVIMA/pembrolizumab were myocardial infarction (3%) and angina pectoris (1%).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageGrade 3 and 4 increased ALT or AST was seen in 9% of patients. Grade ≥2 increased ALT or AST was reported in 64 (18%) patients, of whom 20 (31%) received ≥40 mg daily oral prednisone equivalent. Recurrence of Grade ≥2 increased ALT or AST was observed in 3 patients on rechallenge in patients receiving LENVIMA and 10 patients receiving both LENVIMA and pembrolizumab.
Previously Treated Renal Cell Carcinoma in Combination with Everolimus (Study 205): The safety of LENVIMA was evaluated in Study 205, in which patients with unresectable advanced or metastatic renal cell carcinoma (RCC) were randomized (1:1:1) to LENVIMA 18 mg orally once daily with everolimus 5 mg orally once daily (n=51), LENVIMA 24 mg orally once daily (n=52), or everolimus 10 mg orally once daily (n=50) [see Pharmacology: Pharmacodynamics: Clinical Studies: Renal Cell Carcinoma under Actions]. This data also includes patients on the dose escalation portion of the study who received LENVIMA with everolimus (n=11). The median treatment duration was 8.1 months for LENVIMA with everolimus. Among 62 patients who received LENVIMA with everolimus, the median age was 61 years, 71% were men, and 98% were White.
The most common adverse reactions observed in the LENVIMA with everolimus-treated group (≥30%) were, in order of decreasing frequency, diarrhea, fatigue, arthralgia/myalgia, decreased appetite, vomiting, nausea, stomatitis/oral inflammation, hypertension, peripheral edema, cough, abdominal pain, dyspnea, rash, decreased weight, hemorrhagic events, and proteinuria. The most common serious adverse reactions (≥5%) were renal failure (11%), dehydration (10%), anemia (6%), thrombocytopenia (5%), diarrhea (5%), vomiting (5%), and dyspnea (5%).
Adverse reactions led to dose reductions or interruption in 89% of patients receiving LENVIMA with everolimus. The most common adverse reactions (≥5%) resulting in dose reductions in the LENVIMA with everolimus-treated group were diarrhea (21%), fatigue (8%), thrombocytopenia (6%), vomiting (6%), nausea (5%), and proteinuria (5%).
Treatment discontinuation due to an adverse reaction occurred in 29% of patients in the LENVIMA with everolimus-treated group.
Table 13 presents the adverse reactions in >15% of patients in the LENVIMA with everolimus arm. Study 205 was not designed to demonstrate a statistically significant difference in adverse reaction rates for LENVIMA in combination with everolimus, as compared to everolimus for any specific adverse reaction listed in Table 13. (See Table 13.)
 Click on icon to see table/diagram/image
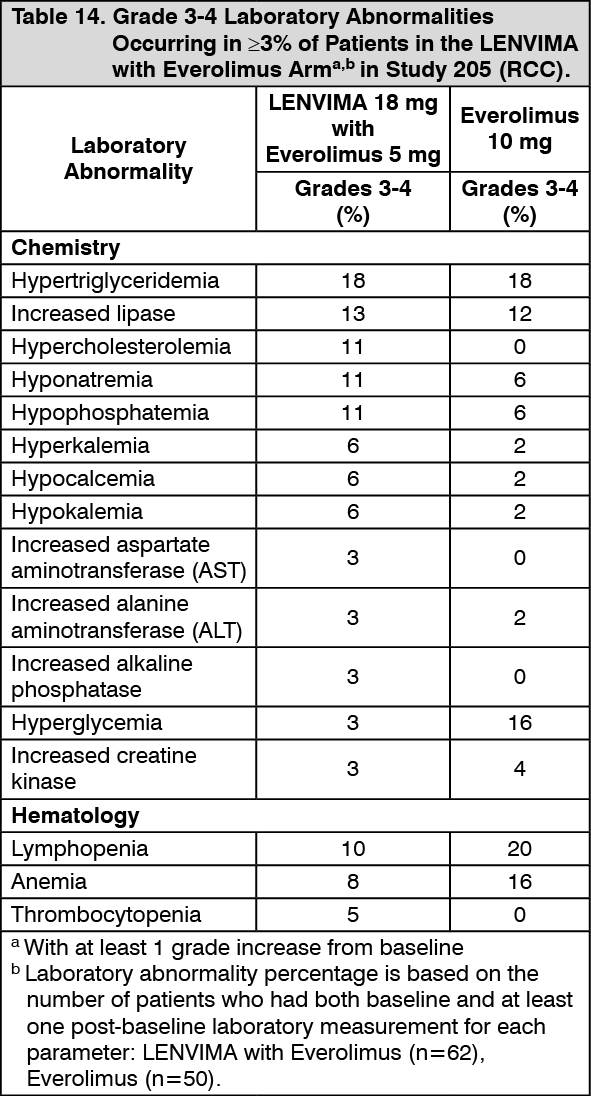
Click on icon to see table/diagram/imageIn Table 14, Grade 3-4 laboratory abnormalities occurring in ≥3% of patients in the LENVIMA with everolimus arm are presented. (See Table 14.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHepatocellular Carcinoma: The safety of LENVIMA was evaluated in REFLECT, which randomized (1:1) patients with unresectable hepatocellular carcinoma (HCC) to LENVIMA (n=476) or sorafenib (n=475) [see Pharmacology: Pharmacodynamics: Clinical Studies: Hepatocellular Carcinoma under Actions]. The dose of LENVIMA was 12 mg orally once daily for patients with a baseline body weight of ≥60 kg and 8 mg orally once daily for patients with a baseline body weight of <60 kg. The dose of sorafenib was 400 mg orally twice daily. Duration of treatment was ≥6 months in 49% and 32% of patients in the LENVIMA and sorafenib groups, respectively. Among the 476 patients who received LENVIMA in REFLECT, the median age was 63 years, 85% were men, 28% were White and 70% were Asian.
The most common adverse reactions observed in the LENVIMA-treated patients (≥20%) were, in order of decreasing frequency, hypertension, fatigue, diarrhea, decreased appetite, arthralgia/myalgia, decreased weight, abdominal pain, palmar-plantar erythrodysesthesia syndrome, proteinuria, dysphonia, hemorrhagic events, hypothyroidism, and nausea.
The most common serious adverse reactions (≥2%) in LENVIMA-treated patients were hepatic encephalopathy (5%), hepatic failure (3%), ascites (3%), and decreased appetite (2%).
Adverse reactions led to dose reduction or interruption in 62% of patients receiving LENVIMA. The most common adverse reactions (≥5%) resulting in dose reduction or interruption of LENVIMA were fatigue (9%), decreased appetite (8%), diarrhea (8%), proteinuria (7%), hypertension (6%), and palmar-plantar erythrodysesthesia syndrome (5%).
Treatment discontinuation due to adverse reactions occurred in 20% of patients in the LENVIMA-treated group. The most common adverse reactions (≥1%) resulting in discontinuation of LENVIMA were fatigue (1%), hepatic encephalopathy (2%), hyperbilirubinemia (1%), and hepatic failure (1%).
Table 15 summarizes the adverse reactions that occurred in ≥10% of patients receiving LENVIMA in REFLECT. REFLECT was not designed to demonstrate a statistically significant reduction in adverse reaction rates for LENVIMA, as compared to sorafenib, for any specified adverse reaction listed in Table 15. (See Table 15.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn Table 16, Grade 3-4 laboratory abnormalities occurring in ≥2% of patients in the LENVIMA arm in REFLECT (HCC) are presented. (See Table 16.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEndometrial Carcinoma: The safety of LENVIMA in combination with pembrolizumab was investigated in Study 309, a multicenter, open-label, randomized (1:1), active-controlled trial in patients with advanced endometrial carcinoma previously treated with at least one prior platinum-based chemotherapy regimen in any setting, including in the neoadjuvant and adjuvant settings [see Pharmacology: Pharmacodynamics: Clinical Studies: Endometrial Carcinoma (EC) under Actions]. Patients with endometrial carcinoma that are pMMR or not MSI-H received LENVIMA 20 mg orally once daily with pembrolizumab 200 mg intravenously every 3 weeks (n=342); or received doxorubicin or paclitaxel (n=325).
For patients with pMMR or not MSI-H status, the median duration of study treatment was 7.2 months (range: 1 day to 26.8 months) and the median duration of exposure to LENVIMA was 6.7 months (range: 1 day to 26.8 months).
Fatal adverse reactions among these patients occurred in 4.7% of those treated with LENVIMA and pembrolizumab, including 2 cases of pneumonia, and 1 case of the following: acute kidney injury, acute myocardial infarction, colitis, decreased appetite, intestinal perforation, lower gastrointestinal hemorrhage, malignant gastrointestinal obstruction, multiple organ dysfunction syndrome, myelodysplastic syndrome, pulmonary embolism, and right ventricular dysfunction.
Serious adverse reactions occurred in 50% of these patients receiving LENVIMA and pembrolizumab. Serious adverse reactions with frequency ≥3% were hypertension (4.4%) and urinary tract infection (3.2%).
Discontinuation of LENVIMA due to an adverse reaction occurred in 26% of these patients. The most common (≥1%) adverse reactions leading to discontinuation of LENVIMA were hypertension (2%), asthenia (1.8%), diarrhea (1.2%), decreased appetite (1.2%), proteinuria (1.2%), and vomiting (1.2%).
Dose reductions of LENVIMA due to adverse reactions occurred in 67% of patients. The most common (≥5%) adverse reactions resulting in dose reduction of LENVIMA were hypertension (18%), diarrhea (11%), palmar-plantar erythrodysesthesia syndrome (9%), proteinuria (7%), fatigue (7%), decreased appetite (6%), asthenia (5%), and weight decreased (5%).
Dose interruptions of LENVIMA due to an adverse reaction occurred in 58% of these patients. The most common (≥2%) adverse reactions leading to interruption of LENVIMA were hypertension (11%), diarrhea (11%), proteinuria (6%), decreased appetite (5%), vomiting (5%), increased alanine aminotransferase (3.5%), fatigue (3.5%), nausea (3.5%), abdominal pain (2.9%), weight decreased (2.6%), urinary tract infection (2.6%), increased aspartate aminotransferase (2.3%), asthenia (2.3%), and palmar-plantar erythrodysesthesia (2%).
Tables 17 and 18 summarize adverse reactions and laboratory abnormalities, respectively, in patients receiving LENVIMA in Study 309. (See Tables 17 and 18.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePost-marketing Experience: The following adverse reactions have been identified during post-approval use of LENVIMA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal: pancreatitis, increased amylase.
General: impaired wound healing.
Hepatobiliary: cholecystitis.
Renal and Urinary: nephrotic syndrome.
Vascular: arterial (including aortic) aneurysms, dissections, and rupture.
View ADR Monitoring Form