Pharmacotherapeutic Group: Other antineoplastic agents.
ATC Code: L01XX41.
Pharmacology: Pharmacodynamics: Mechanism of Action: Eribulin mesilate is a microtubule dynamics inhibitor belonging to the halichondrin class of antineoplastic agents. It is a structurally simplified, synthetic analogue of halichondrin B, a natural product isolated from marine sponge
Halichondria okadai.
Eribulin inhibits the growth phase of microtubules without affecting the shortening phase and sequesters tubulin into non-productive aggregates. Eribulin exerts its effects via a tubulin-based anti-mitotic mechanism leading to G
2/M cell-cycle block, disruption of mitotic spindles and ultimately, apoptotic cell death after prolonged mitotic blockage.
Clinical Efficacy: Breast Cancer: The efficacy of HALAVEN in breast cancer is primarily supported by two randomized Phase 3 comparative studies.
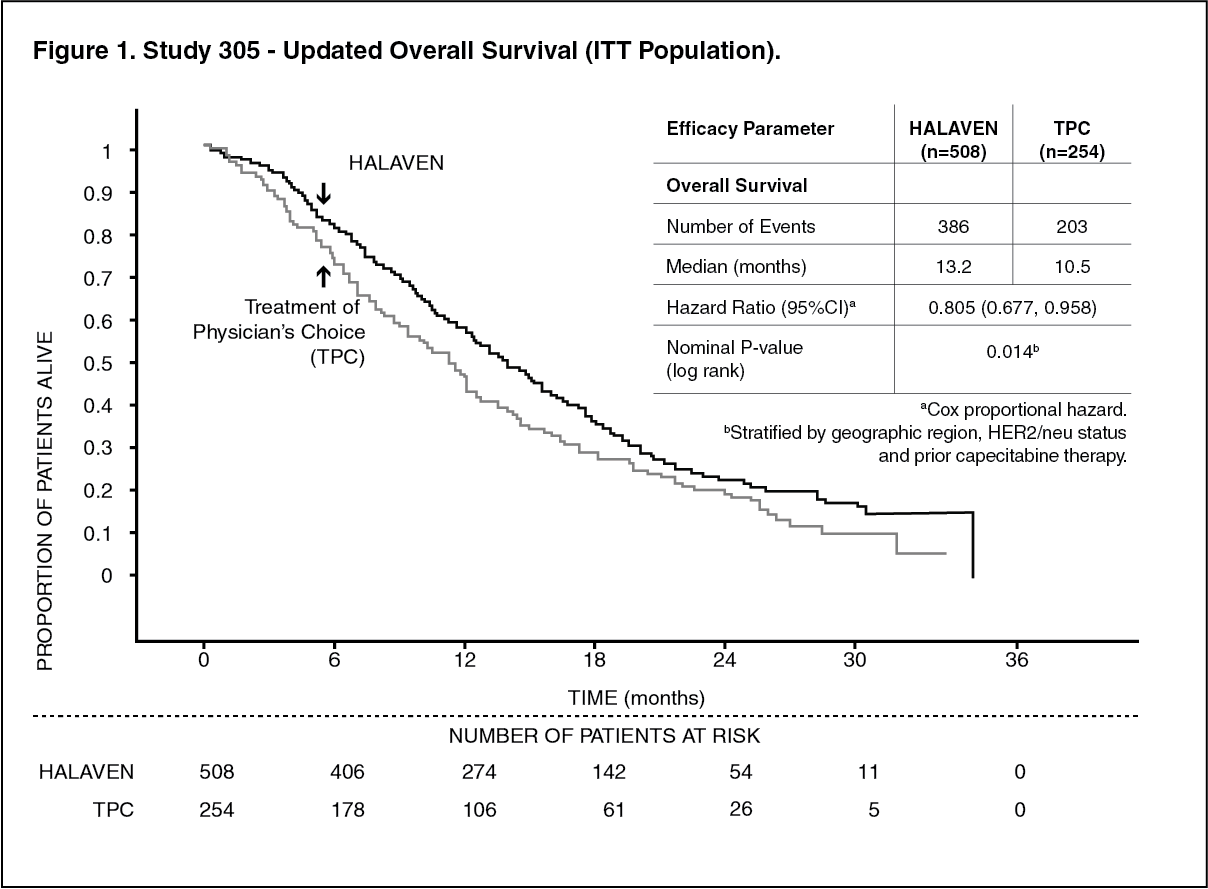
The 762 patients in the pivotal Phase 3 EMBRACE study (Study 305) had locally recurrent or metastatic breast cancer, and had previously received at least two and a maximum of five chemotherapy regimens, including an anthracycline and a taxane (unless contraindicated). Patients must have progressed within 6 months of their last chemotherapeutic regimen. The HER2 status of the patients was 16.1% positive, 74.2% negative and 9.7% unknown, whilst 18.9% of patients were triple negative. They were randomized 2:1 to receive either HALAVEN or treatment of physician's choice (TPC), which consisted of 97% chemotherapy (26% vinorelbine, 18% gemcitabine, 18% capecitabine, 16% taxane, 9% anthracycline, 10% other chemotherapy) or 3% hormonal therapy. The study met its primary endpoint with an overall survival (OS) result that was statistically significantly better in the erbulin group compared to a TPC at 55% of events.
This result was confirmed with an updated overall survival analysis carried out at 77% of events. (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
By independent review, the median progression-free survival (PFS) was 3.7 months for eribulin compared to 2.2 months for the TPC arm (HR 0.865, 95% CI: 0.714, 1.048, p=0.137). In response evaluable patients, the objective response rate by the RECIST criteria was 12.2% (95% CI: 9.4%, 15.5%) by independent review for the eribulin arm compared to 4.7% (95% CI: 2.3%, 8.4%) for the TPC arm.
The positive effect on OS was seen in both taxane-refractory and non-refractory groups of patients. In the OS update, the HR for eribulin versus TPC was 0.9 (95% CI: 0.71, 1.14) in favour of eribulin for taxane-refractory patients and 0.73 (95% CI: 0.56, 0.96) for patients not taxane-refractory.
The positive effect on OS was seen both in capecitabine-naive and in capecitabine pre-treated patient groups. The updated OS analysis showed a survival benefit for the eribulin group compared to TPC both in capecitabine pre-treated patients with a HR of 0.787 (95% CI: 0.645, 0.961), and for the capecitabine-naive patients with a corresponding HR of 0.865 (95% CI: 0.606, 1.233).
The second Phase 3 study in earlier line metastatic breast cancer, Study 301, was an open-label, randomized, study in patients (n=1102) with locally advanced or metastatic breast cancer to investigate the efficacy of HALAVEN monotherapy compared to capecitabine monotherapy in terms of OS and PFS as co-primary endpoint. Patients had previously received up to three prior chemotherapy regimens, including both an anthracycline and a taxane and a maximum of two for advanced disease, with the percentage who had received 0, 1 or 2 prior chemotherapy treatments for metastatic breast cancer being 20%, 52% or 27.2%, respectively. The HER2 status of the patients was: 15.3% positive, 68.5% negative and 16.2% unknown, whilst 25.8% of patients were triple negative. (See Figure 2.)
Click on icon to see table/diagram/image
Progression free survival assessed by independent review was similar between eribulin and capecitabine with medians of 4.1 months versus 4.2 months (HR 1.08 [95% CI: 0.932, 1.250]), respectively. Objective response rate as assessed by independent review was also similar between eribulin and capecitabine; 11% (95% CI: 8.5, 13.9) in the eribulin group and 11.5% (95% CI: 8.9, 14.5) in the capecitabine group.
The overall survival in patients in HER2 negative and HER2 positive patients in the eribulin and control groups in Study 305 and Study 301 are shown in Tables 1 and 2. (See Tables 1 and 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Note: Concomitant anti-HER2 therapy was not included in Study 305 and Study 301.
Liposarcoma: In liposarcoma the efficacy of eribulin is supported by the pivotal Phase 3 sarcoma study (Study 309). The patients in this study (n=452) had locally recurrent, inoperable and/or metastatic soft tissue sarcoma of one of two subtypes - leiomyosarcoma or liposarcoma. Patients had received at least two prior chemotherapy regimens, one of which must have been an anthracycline (unless contraindicated).
Patients must have progressed within 6 months of their last chemotherapeutic regimen. They were randomized 1:1 to receive either eribulin 1.23 mg/m
2 on days 1 and 8 of a 21 day cycle or dacarbazine 850 mg/m
2, 1000 mg/m
2 or 1200 mg/m
2 (dose determined by the investigator prior to randomization), every 21 days.
In Study 309, a statistically significant improvement in OS was observed in patients randomized to the eribulin arm compared to the control arm. This translated into a 2 month improvement in median OS (13.5 months for eribulin treated patients vs. 11.5 months for dacarbazine treated patients). There was no significant difference in progression-free survival or overall response rate between the treatment arms in the overall population.
Treatment effects of eribulin were limited to patients with liposarcoma (45% dedifferentiated, 37% myxoid/round cell and 18% pleomorphic in Study 309) based on pre-planned subgroup analyses of OS and PFS. There was no difference in efficacy between eribulin and dacarbazine in patients with advanced or metastatic leiomyosarcoma. (See Table 3, and Figures 3 and 4.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Paediatric Population: The European Medicines Agency has waived the obligation to submit the results of studies with eribulin in all subsets of the paediatric population in the indication of breast cancer (see Dosage & Administration).
The European Medicines Agency has deferred the obligation to submit the results of studies with HALAVEN in one or more subsets of the paediatric population for the treatment of rhabdomyosarcoma and non-rhabdomyosarcoma soft tissue sarcoma. See Dosage & Administration for information on paediatric use.
Pharmacokinetics: Distribution: The pharmacokinetics of eribulin are characterized by a rapid distribution phase followed by a prolonged elimination phase, with a mean terminal half-life of approximately 40 h. It has a large volume of distribution (range of means 43-114 L/m
2).
Eribulin is weakly bound to plasma proteins. The plasma protein binding of eribulin (100-1000 ng/mL) ranged from 49% to 65% in human plasma.
Biotransformation: Unchanged eribulin was the major circulating species in plasma following administration of
14C-eribulin to patients. Metabolite concentrations represented <0.6% of parent compound, confirming that there are no major human metabolites of eribulin.
Elimination: Eribulin has a low clearance (range of means 1.16-2.42 L/hr/m
2). No significant accumulation of eribulin is observed on weekly administration. The pharmacokinetic properties are not dose or time dependent in the range of eribulin doses of 0.22 to 3.53 mg/m
2.
Eribulin is eliminated primarily by biliary excretion. The transport protein involved in the excretion is presently unknown. Preclinical
in vitro studies indicate that eribulin is transported by Pgp. However it has been shown that at clinically relevant concentrations eribulin is not a Pgp inhibitor
in vitro. Additionally,
in vivo, concomitant administration of ketoconazole, a Pgp inhibitor, has no effect on eribulin exposure (AUC and C
max).
In vitro studies have also indicated that eribulin is not a substrate for OCTI.
After administration of
14C-eribulin to patients, approximately 82% of the dose was eliminated in faeces and 9% in urine indicating that renal clearance is not a significant route of eribulin elimination.
Unchanged eribulin represented most of the total radioactivity in faeces and urine.
Hepatic Impairment: A study evaluated the pharmacokinetics of eribulin in patients with mild (Child-Pugh A; n=7) and moderate (Child-Pugh B; n=4) hepatic impairment due to liver metastases. Compared to patients with normal hepatic function (n=6), eribulin exposure increased 1.8-fold and 3-fold in patients with mild and moderate hepatic impairment, respectively. Administration of HALAVEN at a dose of 0.97 mg/m
2 to patients with mild hepatic impairment and 0.62 mg/m
2 to patients with moderate hepatic impairment resulted in a somewhat higher exposure than after a dose of 1.23 mg/m
2 to patients with normal hepatic function. HALAVEN was not studied in patients with severe hepatic impairment (Child-Pugh C). There is no study in patients with hepatic impairment due to cirrhosis. (See Dosage & Administration.)
Renal Impairment: Increased eribulin exposure was seen in some patients with moderately or severely impaired renal function, with high between-subject variability. The pharmacokinetics of eribulin were evaluated in a Phase 1 study in patients with normal renal function (Creatinine clearance: >80 mL/min; n=6), moderate (30-50 mL/min; n=7) or severe (15-<30 mL/min; n=6) renal impairment. Creatinine clearance was estimated with the Cockcroft-Gault formula. A 1.5-fold (90% CI: 0.9-2.5) higher dose-normalised AUC (
0-inf) was observed in patients with moderate and severe renal impairment. See Dosage & Administration for treatment recommendations.
Toxicology: Preclinical Safety Data: Eribulin was not mutagenic
in vitro in the bacterial reverse mutation assay (Ames test). Eribulin was positive in the mouse lymphoma mutagenesis assay and was clastogenic in the
in vivo rat micronucleus assay.
No carcinogenicity studies have been conducted with eribulin.
A fertility study was not conducted with eribulin, but based on non-clinical findings in repeated-dose studies where testicular toxicity was observed in both rats (hypocellularity of seminiferous epithelium with hypospermia/aspermia) and dogs, male fertility may be compromised by treatment with eribulin. An embryofoetal development study in rat confirmed the development toxicity and teratogenic potential of eribulin. Pregnant rats were treated with eribulin mesilate equivalent to 0.009, 0.027, 0.088 and 0.133 mg/kg eribulin at gestation days 8, 10 and 12. Dose related increased number of resorptions and decreased foetal weight were observed at doses ≥0.088 mg/kg and increased incidence of malformations (absence of lower jaw, tongue, stomach and spleen) was recorded at 0.133 mg/kg.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image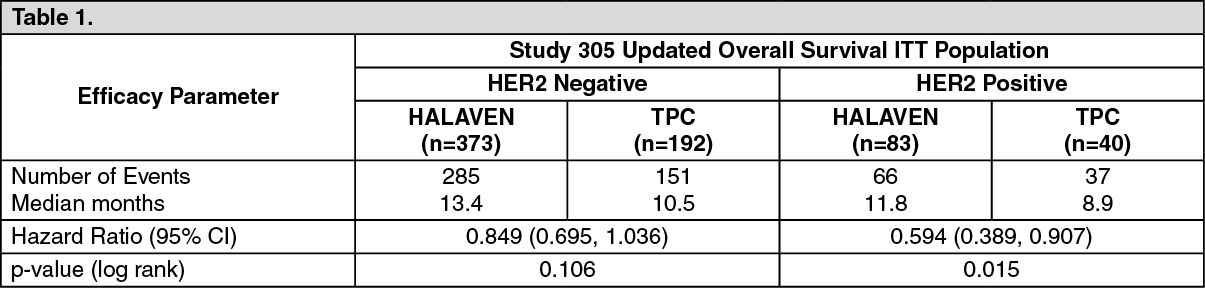 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image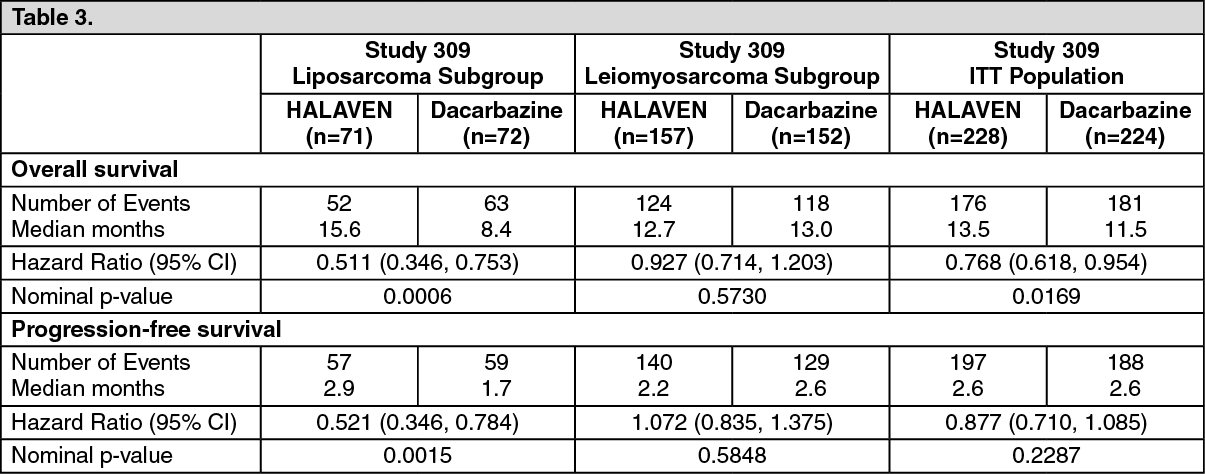 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image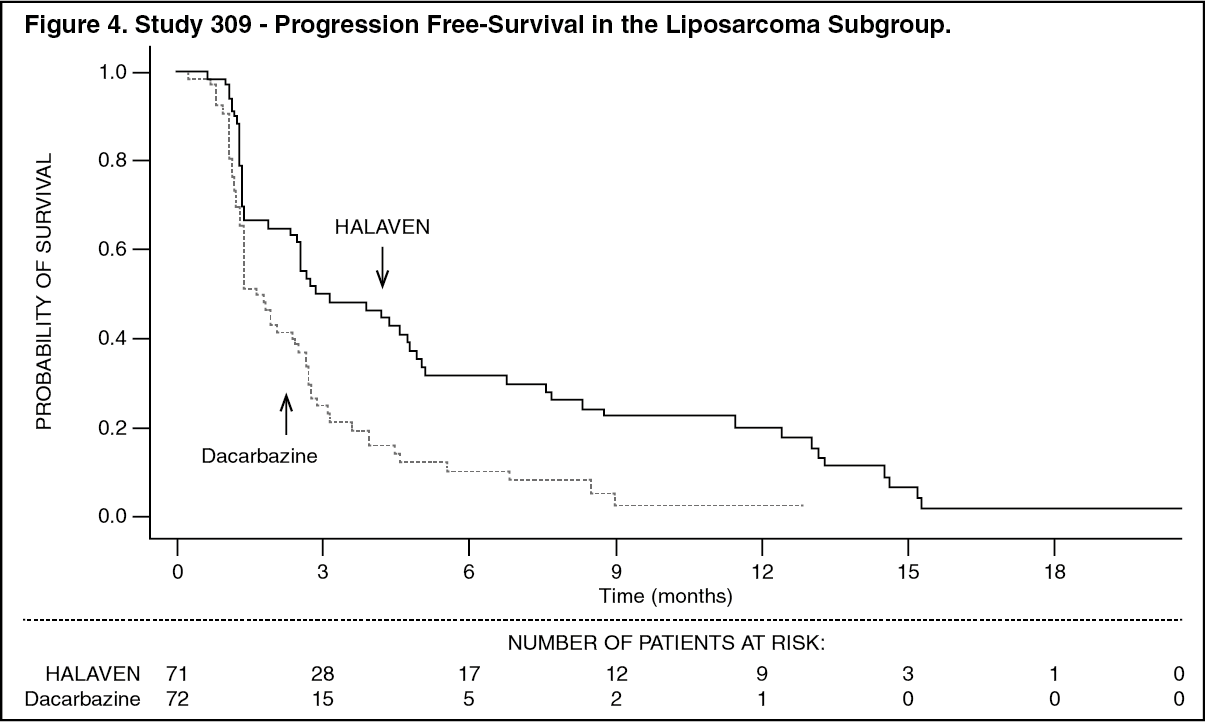 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image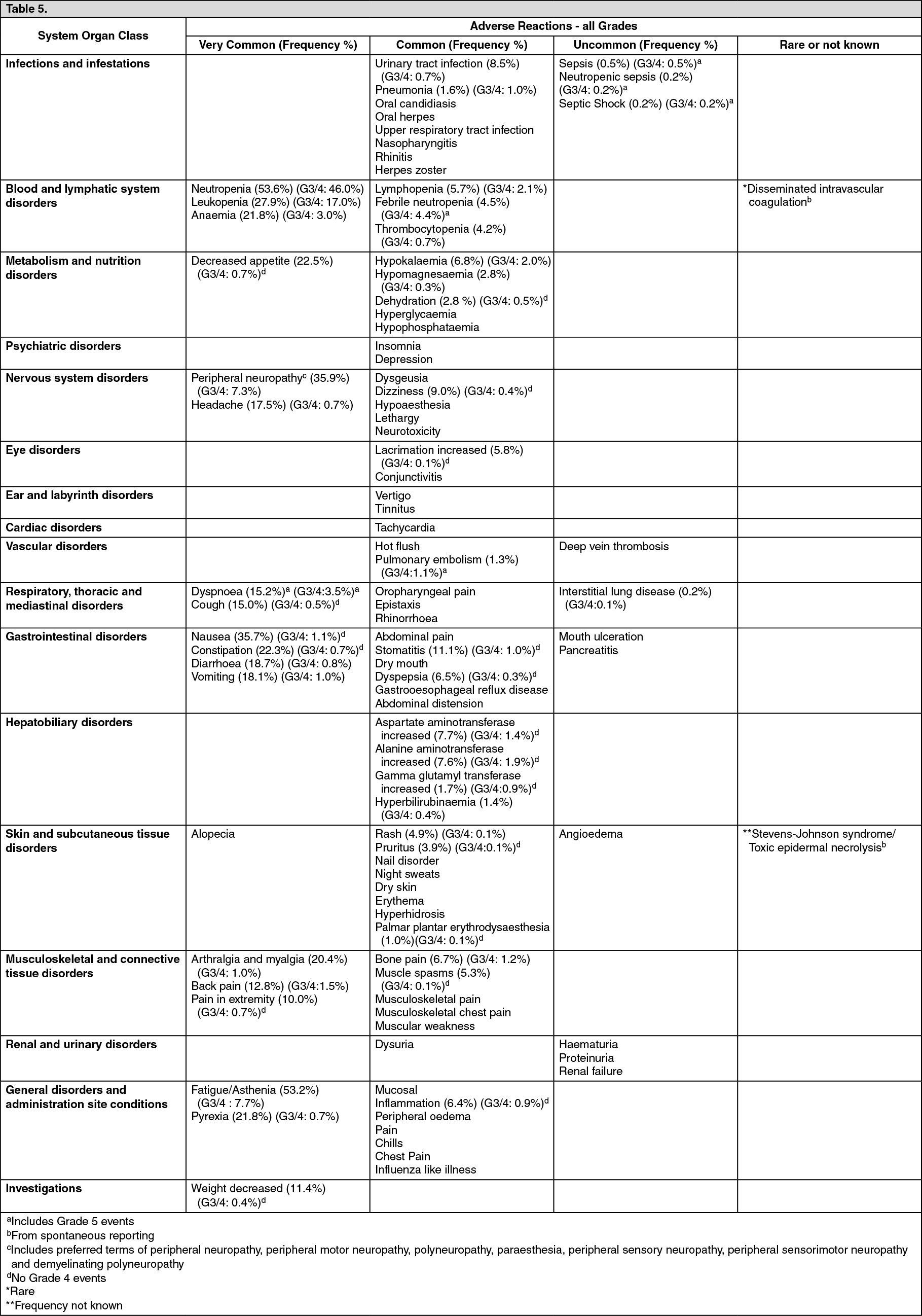 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
