Pharmacotherapeutic group: Platelet aggregation inhibitors excluding heparin.
ATC code: B01AC24.
Pharmacology: Pharmacodynamics: Mechanism of action: Brilinta contains ticagrelor, a member of the chemical class cyclopentyltriazolopyrimidines (CPTP), which is an oral, direct acting, selective and reversibly binding P2Y
12 receptor antagonist that prevents adenosine diphosphate (ADP)-mediated P2Y
12 dependent platelet activation and aggregation. Ticagrelor does not prevent ADP binding but when bound to the P2Y
12 receptor prevents ADP-induced signal transduction. Since platelets participate in the initiation and/or evolution of thrombotic complications of atherosclerotic disease, inhibition of platelet function has been shown to reduce the risk of cardiovascular events such as death, MI or stroke.
Ticagrelor, also increases local endogenous adenosine levels by inhibiting the equilibrative nucleoside transporter-1 (ENT-1).
Ticagrelor has been documented to augment the following adenosine-induced effects in healthy subjects and in patients with ACS: vasodilation (measured by coronary blood flow increases in healthy volunteers and ACS patients; headache), inhibition of platelet function (in human whole blood
in vitro) and dyspnoea. However, a link between the observed increases in adenosine and clinical outcomes (e.g.: morbidity-mortality) has not been clearly elucidated.
Pharmacodynamic effects: Onset of action: In patients with stable coronary artery disease on ASA, ticagrelor demonstrates a rapid onset of pharmacological effect as demonstrated by a mean Inhibition of Platelet Aggregation (IPA) for ticagrelor at 0.5 hours after 180 mg loading dose of about 41%, with the maximum IPA effect of 89% by 2-4 hours post dose, and maintained between 2-8 hours. 90% of patients had final extent IPA >70% by 2 hours post dose.
Offset of action: If a CABG procedure is planned, ticagrelor bleeding risk is increased compared to clopidogrel when discontinued within less than 96 hours prior to procedure.
Switching data: Switching from clopidogrel to ticagrelor results in an absolute IPA increase of 26.4% and switching from ticagrelor to clopidogrel results in an absolute IPA decrease of 24.5%. Patients can be switched from clopidogrel to ticagrelor without any interruption of antiplatelet effect (see Dosage & Administration).
Clinical efficacy and safety: The clinical evidence for the efficacy and safety of ticagrelor is derived from two phase 3 trials: The PLATO [PLATelet Inhibition and Patient Outcomes] study, a comparison of ticagrelor to clopidogrel, both given in combination with ASA and other standard therapy.
The PEGASUS TIMI-54 [PrEvention with TicaGrelor of SecondAry Thrombotic Events in High-RiSk AcUte Coronary Syndrome Patients] study, a comparison of ticagrelor combined with ASA to ASA therapy alone.
PLATO study (Acute Coronary Syndromes): The PLATO study included 18,624 patients who presented within 24 hours of onset of symptoms of unstable angina (UA), non ST elevation myocardial infarction (NSTEMI) or ST elevation myocardial infarction (STEMI), and were initially managed medically, or with percutaneous coronary intervention (PCI), or with coronary artery bypass grafting (CABG).
Clinical efficacy: On a background of daily ASA, ticagrelor 90 mg twice daily showed superiority to 75 mg daily clopidogrel in preventing the composite endpoint of CV death, MI, or stroke, with the difference driven by CV death and MI. Patients received a 300 mg loading dose of clopidogrel (600 mg possible if having PCI) or 180 mg of ticagrelor.
The result appeared early (absolute risk reduction [ARR] 0.6% and Relative Risk Reduction [RRR] of 12% at 30 days), with a constant treatment effect over the entire 12 month period, yielding ARR 1.9% per year with RRR of 16%. This suggests it is appropriate to treat patients with ticagrelor 90 mg twice daily for 12 months (see Dosage & Administration). Treating 54 ACS patients with ticagrelor instead of clopidogrel will prevent 1 atherothrombotic event; treating 91 will prevent 1 CV death (see Figure 1 and Table 1).
The treatment effect of ticagrelor over clopidogrel appears consistent across many subgroups, including weight; sex; medical history of diabetes mellitus, transient ischaemic attack or non-haemorrhagic stroke, or revascularisation; concomitant therapies including heparins, GpIIb/IIIa inhibitors and proton pump inhibitors (see Interactions); final index event diagnosis (STEMI, NSTEMI, or UA); and, treatment pathway intended at randomisation (invasive or medical).
A weakly significant treatment interaction was observed with region whereby the hazard ratio (HR) for the primary endpoint favours ticagrelor in the rest of world but favours clopidogrel in North America, which represented approximately 10% of the overall population studied (interaction p-value=0.045). Exploratory analyses suggest a possible association with ASA dose such that reduced efficacy was observed with ticagrelor with increasing ASA doses. Chronic daily ASA doses to accompany Brilinta should be 75-150 mg (see Dosage & Administration and Precautions).
Figure 1 shows the estimate of the risk to the first occurrence of any event in the composite efficacy endpoint. (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Ticagrelor reduced the occurrence of the primary composite endpoint compared to clopidogrel in both the UA/NSTEMI and STEMI population (Table 1). Thus, Brilinta 90 mg twice daily together with low-dose ASA can be used in patients with ACS (unstable angina, non ST elevation Myocardial Infarction [NSTEMI] or ST elevation Myocardial Infarction [STEMI]); including patients managed medically, and those who are managed with percutaneous coronary intervention (PCI) or coronary artery by-pass grafting (CABG). (See Table 1.)
Click on icon to see table/diagram/image
PLATO genetic substudy: CYP2C19 and ABCB1 genotyping of 10,285 patients in PLATO provided associations of genotype groups with PLATO outcomes. The superiority of ticagrelor over clopidogrel in reducing major CV events was not significantly affected by patient CYP2C19 or ABCB1 genotype. Similar to the overall PLATO study, total PLATO Major bleeding did not differ between ticagrelor and clopidogrel, regardless of CYP2C19 or ABCB1 genotype. Non-CABG PLATO Major bleeding was increased with ticagrelor compared clopidogrel in patients with one or more CYP2C19 loss of function alleles, but similar to clopidogrel in patients with no loss of function allele.
Combined efficacy and safety composite: A combined efficacy and safety composite (CV death, MI, stroke, or PLATO-defined 'Total Major' bleeding) indicates that the benefit in efficacy of ticagrelor compared to clopidogrel is not offset by the major bleeding events (ARR 1.4%, RRR 8%, HR 0.92; p=0.0257) over 12 months after ACS.
Clinical safety: Holter substudy: To study the occurrence of ventricular pauses and other arrhythmic episodes during PLATO, investigators performed Holter monitoring in a subset of nearly 3000 patients, of whom approximately 2000 had recordings both in the acute phase of their ACS and after one month. The primary variable of interest was the occurrence of ventricular pauses ≥3 seconds. More patients had ventricular pauses with ticagrelor (6.0%) than with clopidogrel (3.5%) in the acute phase; and 2.2% and 1.6% respectively after 1 month (see Precautions). The increase in ventricular pauses in the acute phase of ACS was more pronounced in ticagrelor patients with history of CHF (9.2% versus 5.4% in patients without CHF history; for clopidogrel patients, 4.0% in those with versus 3.6% in those without CHF history). This imbalance did not occur at one month: 2.0% versus 2.1% for ticagrelor patients with and without CHF history respectively; and 3.8% versus 1.4% with clopidogrel. There were no adverse clinical consequences associated with this imbalance (including pacemaker insertions) in this population of patients.
PEGASUS study (History of Myocardial Infarction): The PEGASUS TIMI-54 study was a 21,162 patient, event-driven, randomised, double-blind, placebo-controlled, parallel group, international multicentre study to assess the prevention of atherothrombotic events with ticagrelor given at 2 doses (either 90 mg twice daily or 60 mg twice daily) combined with low dose ASA (75-150 mg), compared to ASA therapy alone in patients with history of MI and additional risk factors for atherothrombosis.
Patients were eligible to participate if they were aged 50 years or over, with a history of MI (1 to 3 years prior to randomisation), and had at least one of the following risk factors for atherothrombosis: age ≥65 years, diabetes mellitus requiring medication, a second prior MI, evidence of multivessel CAD, or chronic non-end-stage renal dysfunction.
Patients were ineligible if there was planned use of a P2Y12 receptor antagonist, dipyridamole, cilostazol, or anticoagulant therapy during the study period; if they had a bleeding disorder or a history of an ischemic stroke or intracranial bleeding, a central nervous system tumour, or an intracranial vascular abnormality; if they had had gastrointestinal bleeding within the previous 6 months or major surgery within the previous 30 days.
Clinical efficacy: (See Figure 2 and Table 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Both 60 mg twice daily and 90 mg twice daily regimens of ticagrelor in combination with ASA were superior to ASA alone in the prevention of atherothrombotic events (composite endpoint: CV death, MI and stroke), with a consistent treatment effect over the entire study period, yielding a 16% RRR and 1.27% ARR for ticagrelor 60 mg and a 15% RRR and 1.19% ARR for ticagrelor 90 mg.
Although the efficacy profiles of 90 mg and 60 mg were similar, there is evidence that the lower dose has a better tolerability and safety profile in relation to risk of the bleeding and dyspnoea. Therefore only Brilinta 60 mg twice daily co-administered with ASA is recommended for the prevention atherothrombotic events (CV death, MI and stroke) in patients with a history of MI and a high risk of developing an atherothrombotic event.
Relative to ASA alone, ticagrelor 60 mg twice daily significantly reduced the primary composite endpoint of CV death, MI and stroke. Each of the components contributed to the reduction in the primary composite endpoint (CV death 17% RRR, MI 16% RRR, and stroke 25% RRR).
The RRR for the composite endpoint from 1 to 360 days (17% RRR) and from 361 days and onwards (16% RRR) was similar. There are limited data on the efficacy and safety of ticagrelor beyond 3 years of extended treatment.
There was no evidence of benefit (no reduction in the primary composite endpoint of CV death, MI and stroke, but an increase in major bleeding) when ticagrelor 60 mg twice daily was introduced in clinically stable patients >2 years from the MI, or more than one year after stopping previous ADP receptor inhibitor treatment (see Dosage & Administration).
Clinical safety: The rate of discontinuations with ticagrelor 60 mg due to bleeding and dyspnoea was higher in patients >75 years (42%) than in younger patients (range: 23-31%), with a difference versus placebo higher than 10% (42% vs. 29%) in patients >75 years.
Paediatric population: See Dosage & Administration for information on paediatric use.
Pharmacokinetics: Ticagrelor demonstrates linear pharmacokinetics and exposure to ticagrelor and the active metabolite (AR-C124910XX) are approximately dose proportional up to 1260 mg.
Absorption: Absorption of ticagrelor is rapid with a median t
max of approximately 1.5 hours. The formation of the major circulating metabolite AR-C124910XX (also active) from ticagrelor is rapid with a median t
max of approximately 2.5 hours. Following oral administration of ticagrelor 90 mg single dose under fasted conditions, C
max is 529 ng/ml and AUC is 3451 ng*h/ml. The metabolite parent ratios are 0.28 for C
max and 0.42 for AUC. The metabolite parent ratios are 0.28 for C
max and 0.42 for AUC. The pharmacokinetics of ticagrelor and AR-C124910XX in patients with a history of MI were generally similar to that in the ACS population. Based on a population pharmacokinetic analysis of the PEGASUS study the median ticagrelor C
max was 391 ng/ml and AUC was 3801 ng*h/ml at steady state for ticagrelor 60 mg. For ticagrelor 90 mg C
max was 627 ng/ml and AUC was 6255 ng*h/ml at steady state.
The mean absolute bioavailability of ticagrelor was estimated to be 36%. Ingestion of a high-fat meal resulted in a 21% increase in ticagrelor AUC and 22% decrease in the active metabolite C
max but had no effect on ticagrelor C
max or the AUC of the active metabolite. These small changes are considered of minimal clinical significance; therefore, ticagrelor can be given with or without food. Ticagrelor as well as the active metabolite are P-gp substrates.
Ticagrelor as crushed tablets mixed in water, given orally or administered through a nasogastric tube into the stomach, has a comparable bioavailability to whole tablets with regards to AUC and C
max for ticagrelor and the active metabolite. Initial exposure (0.5 and 1 hour post-dose) from crushed ticagrelor tablets mixed in water was higher compared to whole tablets, with a generally identical concentration profile thereafter (2 to 48 hours).
Distribution: The steady state volume of distribution of ticagrelor is 87.5 l. Ticagrelor and the active metabolite is extensively bound to human plasma protein (>99. 0%).
Biotransformation: CYP3A4 is the major enzyme responsible for ticagrelor metabolism and the formation of the active metabolite and their interactions with other CYP3A substrates ranges from activation through to inhibition.
The major metabolite of ticagrelor is AR-C124910XX, which is also active as assessed by
in vitro binding to the platelet P2Y
12 ADP-receptor. The systemic exposure to the active metabolite is approximately 30-40% of that obtained for ticagrelor.
Elimination: The primary route of ticagrelor elimination is via hepatic metabolism. When radiolabeled ticagrelor is administered, the mean recovery of radioactivity is approximately 84% (57.8% in faeces, 26.5% in urine). Recoveries of ticagrelor and the active metabolite in urine were both less than 1% of the dose. The primary route of elimination for the active metabolite is most likely via biliary secretion. The mean t
1/2 was approximately 7 hours for ticagrelor and 8.5 hours for the active metabolite.
Special populations: Elderly: Higher exposures to ticagrelor (approximately 25% for both C
max and AUC) and the active metabolite were observed in elderly (≥ 75years) ACS patients compared to younger patients by the population pharmacokinetic analysis. These differences are not considered clinically significant (see Dosage & Administration).
Paediatric: Ticagrelor has not been evaluated in a paediatric population (see Dosage & Administration and Pharmacology: Pharmacodynamics previously mentioned).
Gender: Higher exposures to ticagrelor and the active metabolite were observed in women compared to men. These differences are not considered clinically significant.
Renal impairment: Exposure to ticagrelor were approximately 20% lower and exposure to the active metabolite was approximately 17% higher in patients with severe renal impairment (creatinine clearance <30 ml/min) compared to subjects with normal renal function (see Dosage & Administration).
Hepatic impairment: C
max and AUC for ticagrelor were 12% and 23% higher in patients with mild hepatic impairment compared to matched healthy subjects, respectively, however, the IPA effect of ticagrelor was similar between the two groups. No dose adjustment is needed for patients with mild hepatic impairment. Ticagrelor has not been studied in patients with severe hepatic impairment and there is no pharmacokinetic information in patients with moderate hepatic impairment. In patients that had moderate or severe elevation in one or more liver function tests at baseline, ticagrelor plasma concentrations were on average similar or slightly higher as compared to those without baseline elevations. No dose adjustment is recommended in patients with moderate hepatic impairment (see Contraindications and Precautions).
Ethnicity: Patients of Asian descent have a 39% higher mean bioavailability compared to Caucasian patients. Patients self-identified as Black had an 18% lower bioavailability of ticagrelor compared to Caucasian patients. In clinical pharmacology studies, the exposure (C
max and AUC) to ticagrelor in Japanese subjects was approximately 40% (20% after adjusting for body weight) higher compared to that in Caucasians. The exposure in patients self-identified as Hispanic or Latino was similar to that in Caucasians.
Toxicology: Preclinical safety data: Preclinical data for ticagrelor and its major metabolite have not demonstrated unacceptable risk for adverse effects for humans based on conventional studies of safety pharmacology, single and repeated dose toxicity and genotoxic potential.
Gastrointestinal irritation was observed in several animal species at clinical relevant exposure levels (see Adverse Reactions).
In female rats, ticagrelor at high dose showed an increased incidence of uterine tumors (adenocarcinomas) and an increased incidence of hepatic adenomas. The mechanism for uterine tumors is likely hormonal imbalance which can lead to tumors in rats. The mechanism for the hepatic adenomas is likely due to a rodent-specific enzyme induction in the liver. Thus, the carcinogenicity findings are considered unlikely to be relevant for humans.
In rats minor developmental anomalies were seen at a maternal toxic dose (safety margin of 5.1). In rabbits a slight delay in hepatic maturity and skeletal development was seen in foetuses from dams at high dose without showing maternal toxicity (safety margin of 4.5).
Studies in rats and rabbits have shown reproductive toxicity, with slightly reduced maternal body weight gain and reduced neonatal viability and birth weight, with delayed growth. Ticagrelor produced irregular cycles (mostly extended cycles) in female rats, but did not affect overall fertility in male and female rats. Pharmacokinetic studies performed with radio-labeled ticagrelor have shown that the parent compound and its metabolites are excreted in the milk of rats (see Use in Pregnancy & Lactation).




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image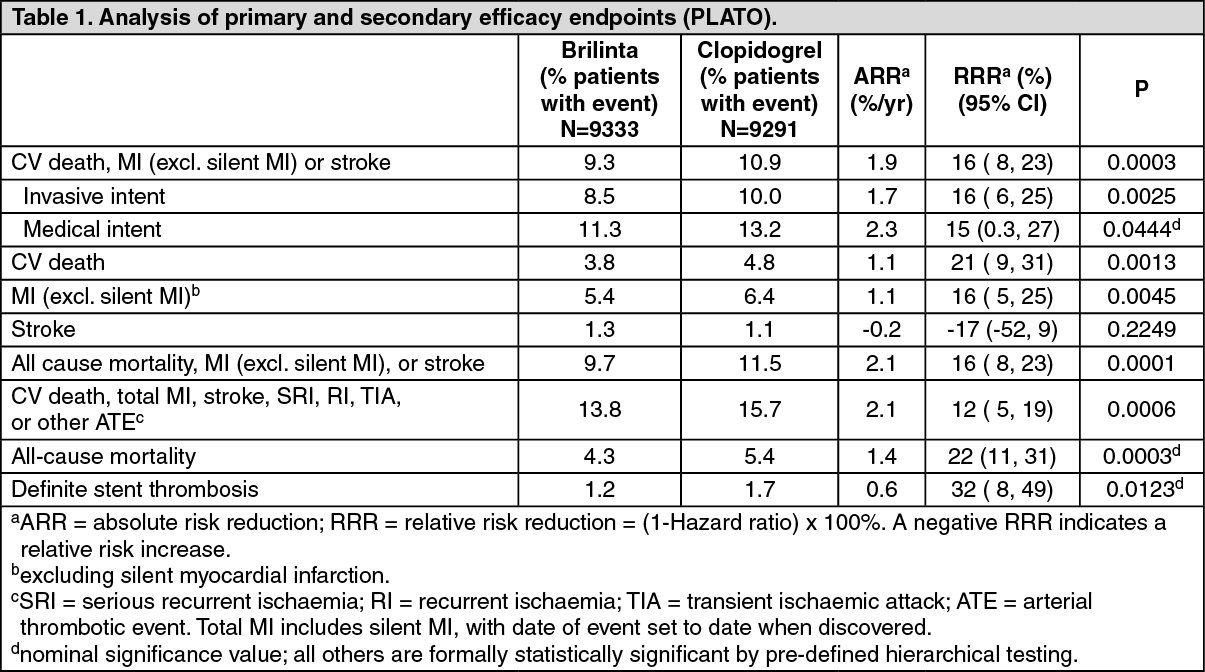 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image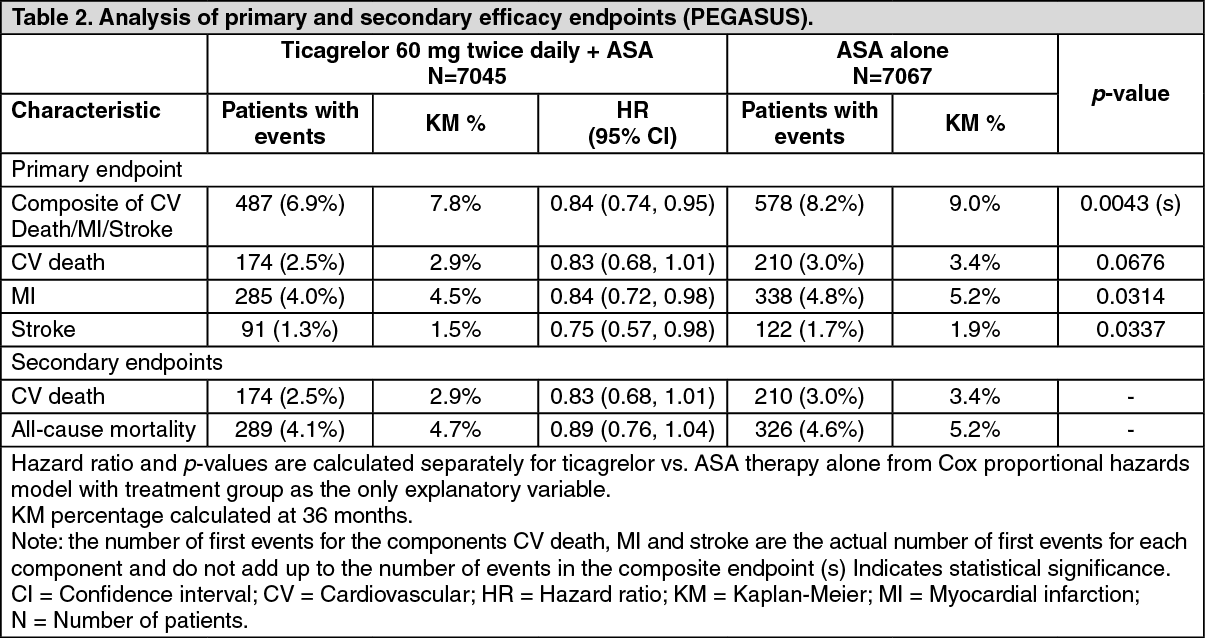 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image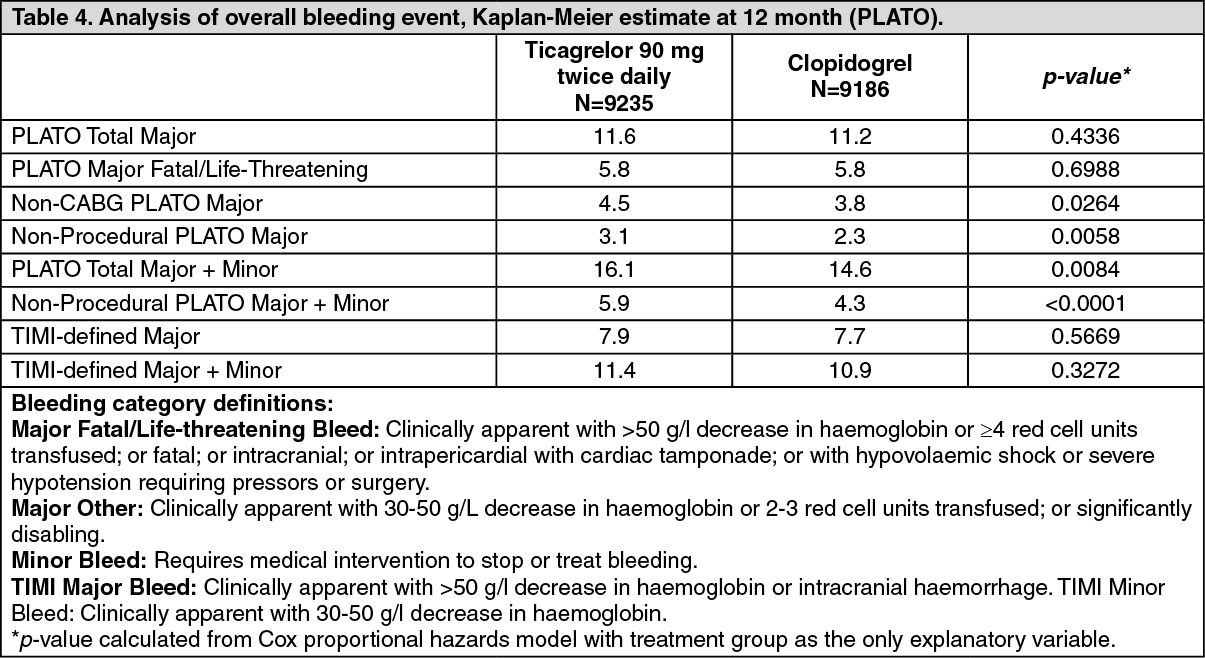 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image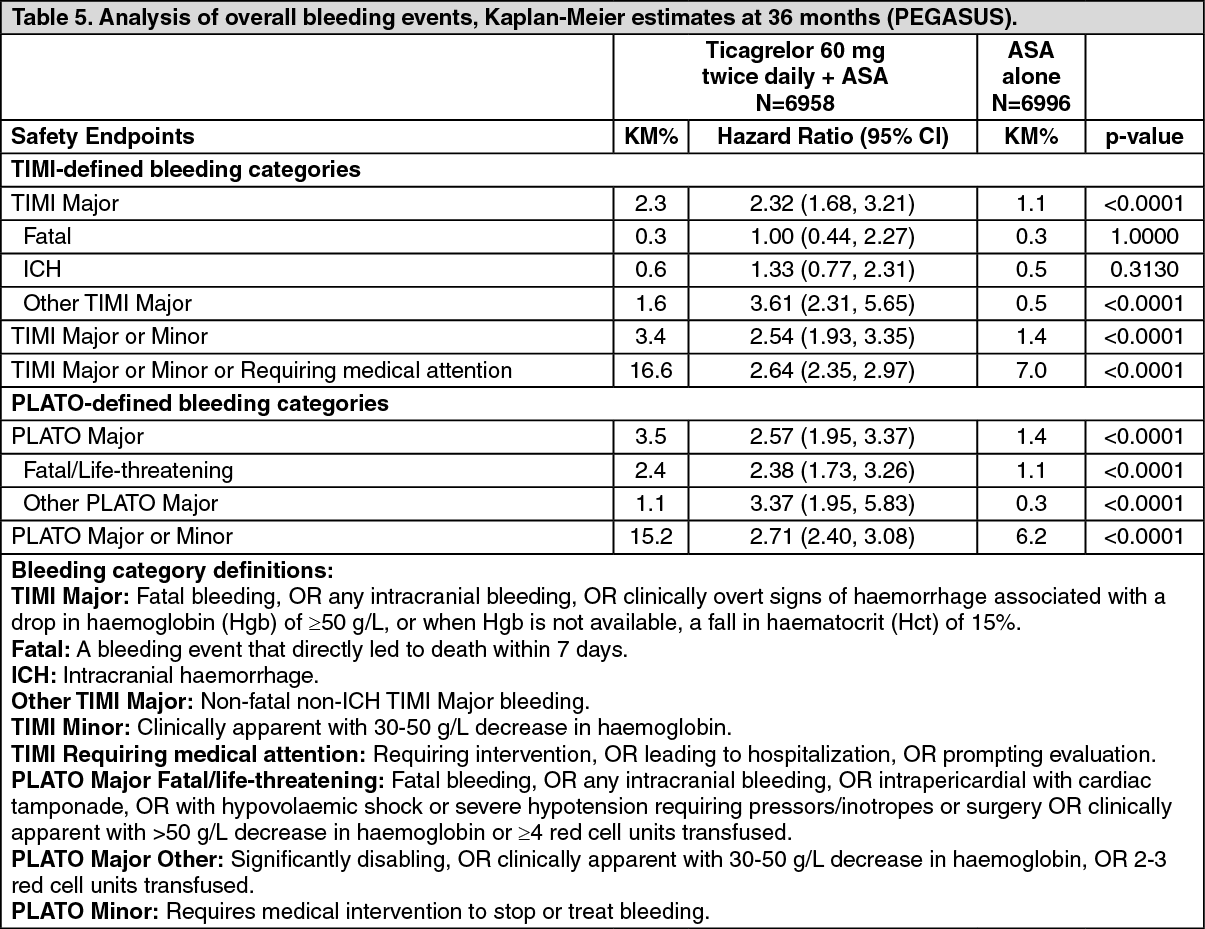 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
