


 Sign Out
Sign Out



Pharmacology: Pharmacodynamics: Mechanism of action: Rivaroxaban is a highly selective direct factor Xa inhibitor with oral bioavailability. Inhibition of factor Xa interrupts the intrinsic and extrinsic pathway of the blood coagulation cascade, inhibiting both thrombin formation and development of thrombi. Rivaroxaban does not inhibit thrombin (activated Factor II) and no effects on platelets have been demonstrated.
Pharmacodynamic effects: Dose-dependent inhibition of factor Xa activity was observed in humans. Prothrombin time (PT) is influenced by rivaroxaban in a dose dependent way with a close correlation to plasma concentrations (r value equals 0.98) if Neoplastin is used for the assay. Other reagents would provide different results. The readout for PT is to be done in seconds, because the INR (International Normalised Ratio) is only calibrated and validated for coumarins and cannot be used for any other anticoagulant.
In patients receiving rivaroxaban for treatment of DVT and PE and prevention of recurrence, the 5/95 percentiles for PT (Neoplastin) 2 - 4 hours after tablet intake (i.e. at the time of maximum effect) for 15 mg rivaroxaban twice daily ranged from 17 to 32 s and for 20 mg rivaroxaban once daily from 15 to 30 s. At trough (8 - 16 h after tablet intake) the 5/95 percentiles for 15 mg twice daily ranged from 14 to 24 s and for 20 mg once daily (18 - 30 h after tablet intake) from 13 to 20 s.
In patients with non-valvular atrial fibrillation receiving rivaroxaban for the prevention of stroke and systemic embolism, the 5/95 percentiles for PT (Neoplastin) 1 - 4 hours after tablet intake (i.e. at the time of maximum effect) in patients treated with 20 mg once daily ranged from 14 to 40 s and in patients with moderate renal impairment treated with 15 mg once daily from 10 to 50 s. At trough (16 - 36 h after tablet intake) the 5/95 percentiles in patients treated with 20 mg once daily ranged from 12 to 26 s and in patients with moderate renal impairment treated with 15 mg once daily from 12 to 26 s.
In a clinical pharmacology study on the reversal of rivaroxaban pharmacodynamics in healthy adult subjects (n=22), the effects of single doses (50 IU/kg) of two different types of PCCs, a 3-factor PCC (Factors II, IX and X) and a 4-factor PCC (Factors II, VII, IX and X) were assessed. The 3-factor PCC reduced mean Neoplastin PT values by approximately 1.0 second within 30 minutes, compared to reductions of approximately 3.5 seconds observed with the 4-factor PCC. In contrast, the 3-factor PCC had a greater and more rapid overall effect on reversing changes in endogenous thrombin generation than the 4-factor PCC (see Overdose).
The activated partial thromboplastin time (aPTT) and HepTest are also prolonged dose-dependently; however, they are not recommended to assess the pharmacodynamic effect of rivaroxaban. There is no need for monitoring of coagulation parameters during treatment with rivaroxaban in clinical routine. However, if clinically indicated rivaroxaban levels can be measured by calibrated quantitative antifactor Xa tests (see Pharmacokinetics as follows).
Clinical efficacy and safety: Prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation: The Xarelto clinical program was designed to demonstrate the efficacy of Xarelto for the prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation.
In the pivotal double-blind ROCKET AF study, 14,264 patients were assigned either to Xarelto 20 mg once daily (15 mg once daily in patients with creatinine clearance 30 - 49 ml/min) or to warfarin titrated to a target INR of 2.5 (therapeutic range 2.0 to 3.0). The median time on treatment was 19 months and overall treatment duration was up to 41 months.
34.9% of patients were treated with acetylsalicylic acid and 11.4% were treated with class III antiarrhythmic including amiodarone.
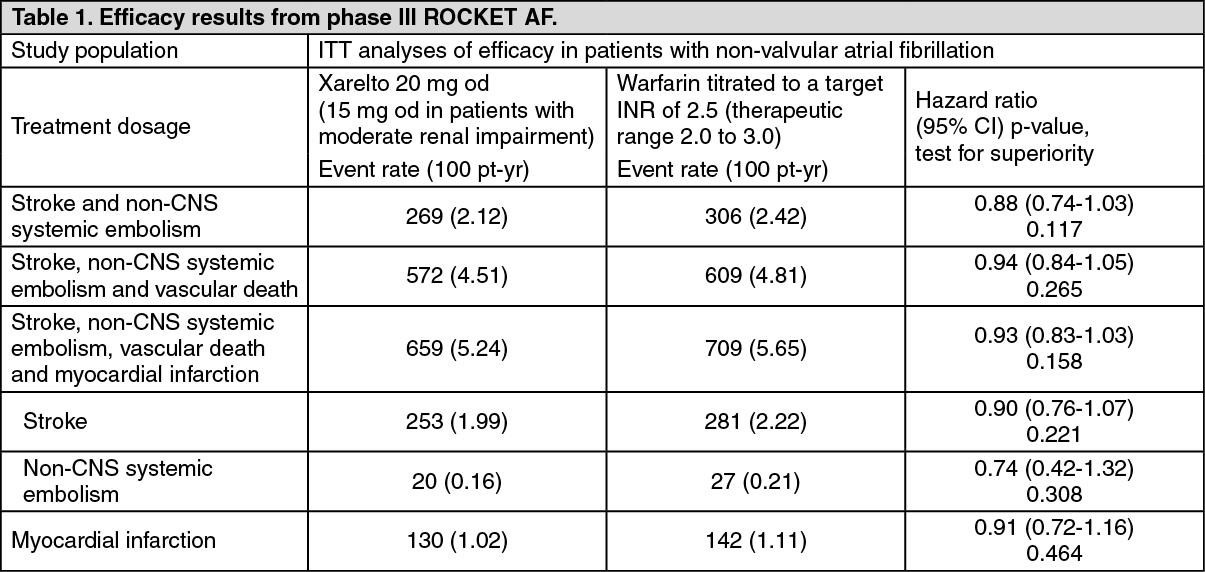
Xarelto was non-inferior to warfarin for the primary composite endpoint of stroke and non-CNS systemic embolism. In the per-protocol population on treatment, stroke or systemic embolism occurred in 188 patients on rivaroxaban (1.71% per year) and 241 on warfarin (2.16% per year) (HR 0.79; 95% CI, 0.66 - 0.96; P<0.001 for non-inferiority). Among all randomised patients analysed according to ITT, primary events occurred in 269 on rivaroxaban (2.12% per year) and 306 on warfarin (2.42% per year) (HR 0.88; 95% CI, 0.74 - 1.03; P<0.001 for non-inferiority; P=0.117 for superiority). Results for secondary endpoints as tested in hierarchical order in the ITT analysis are displayed in Table 1.
Among patients in the warfarin group, INR values were within the therapeutic range (2.0 to 3.0) a mean of 55% of the time (median, 58%; interquartile range, 43 to 71). The effect of rivaroxaban did not differ across the level of centre TTR (Time in Target INR Range of 2.0 - 3.0) in the equally sized quartiles (P=0.74 for interaction). Within the highest quartile according to centre, the hazard ratio with rivaroxaban versus warfarin was 0.74 (95% CI, 0.49 - 1.12).
The incidence rates for the principal safety outcome (major and non-major clinically relevant bleeding events) were similar for both treatment groups (see Table 2). (See Table 1 and Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image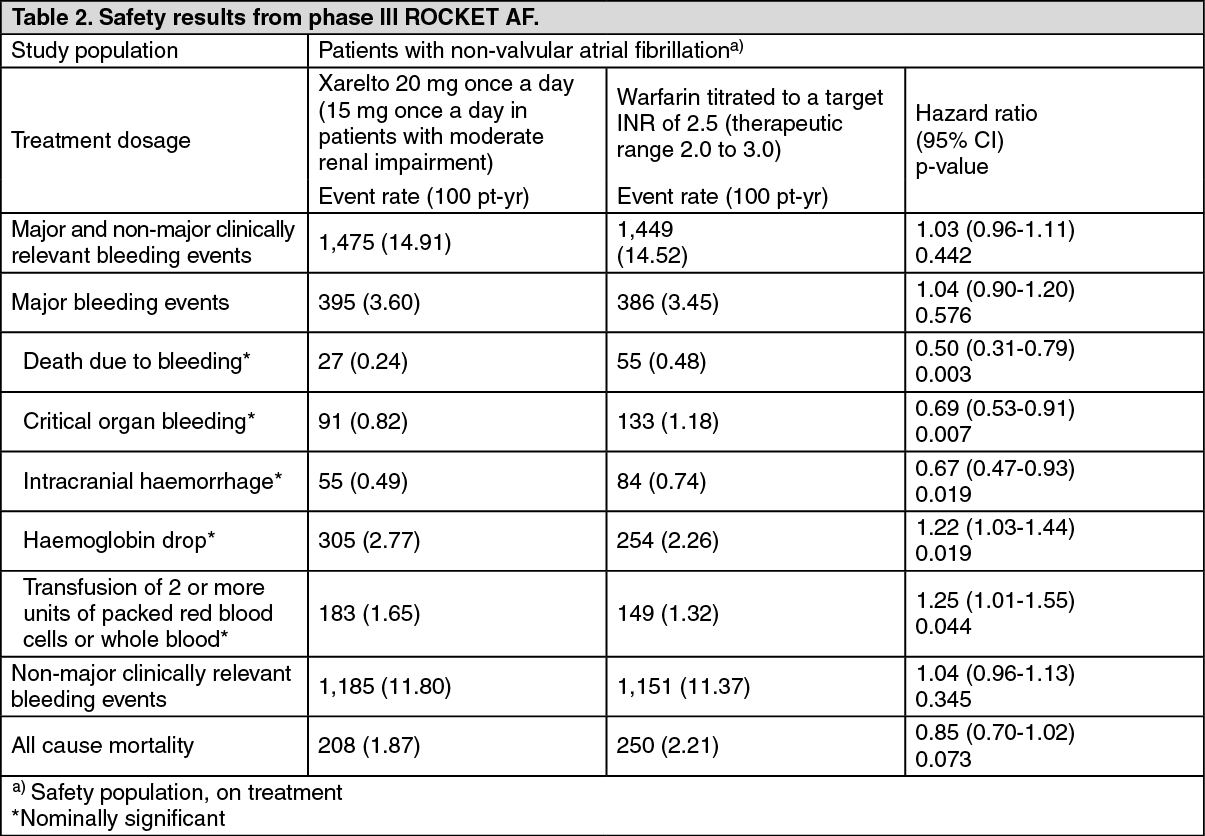 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients undergoing cardioversion: A prospective, randomized, open-label, multicenter, exploratory study with blinded endpoint evaluation (X-VERT) was conducted in 1504 patients (oral anticoagulant naive and pre-treated) with non-valvular atrial fibrillation scheduled for cardioversion to compare rivaroxaban with dose-adjusted VKA (randomized 2:1), for the prevention of cardiovascular events. TEE- guided (1 - 5 days of pretreatment) or conventional cardioversion (at least three weeks of pre-treatment) strategies were employed. The primary efficacy outcome (all stroke, transient ischemic attack, non-CNS systemic embolism, MI and cardiovascular death) occurred in 5 (0.5%) patients in the rivaroxaban group (n = 978) and 5 (1.0%) patients in the VKA group (n = 492; RR 0.50; 95% CI 0.15-1.73; modified ITT population). The principal safety outcome (major bleeding) occurred in 6 (0.6%) and 4 (0.8%) patients in the rivaroxaban (n = 988) and VKA (n = 499) groups, respectively (RR 0.76; 95% CI 0.21- 2.67; safety population). This exploratory study showed comparable efficacy and safety between rivaroxaban and VKA treatment groups in the setting of cardioversion.
Treatment of DVT, PE and prevention of recurrent DVT and PE: The Xarelto clinical program was designed to demonstrate the efficacy of Xarelto in the initial and continued treatment of acute DVT and PE and prevention of recurrence.
Over 9,400 patients were studied in three randomised controlled phase III clinical studies (Einstein DVT, Einstein PE and Einstein Extension) and additionally a predefined pooled analysis of the Einstein DVT and Einstein PE studies was conducted. The overall combined treatment duration in all studies was up to 21 months.
In Einstein DVT 3,449 patients with acute DVT were studied for the treatment of DVT and the prevention of recurrent DVT and PE (patients who presented with symptomatic PE were excluded from this study). The treatment duration was for 3, 6 or 12 months depending on the clinical judgement of the investigator.
For the initial 3 week treatment of acute DVT 15 mg rivaroxaban was administered twice daily. This was followed by 20 mg rivaroxaban once daily.
In Einstein PE, 4,832 patients with acute PE were studied for the treatment of PE and the prevention of recurrent DVT and PE. The treatment duration was for 3, 6 or 12 months depending on the clinical judgement of the investigator.
For the initial treatment of acute PE 15 mg rivaroxaban was administered twice daily for three weeks. This was followed by 20 mg rivaroxaban once daily.
In both the Einstein DVT and the Einstein PE study, the comparator treatment regimen consisted of enoxaparin administered for at least 5 days in combination with vitamin K antagonist treatment until the PT/INR was in therapeutic range (≥ 2.0). Treatment was continued with a vitamin K antagonist dose-adjusted to maintain the PT/INR values within the therapeutic range of 2.0 to 3.0.
In Einstein Extension 1,197 patients with DVT or PE were studied for the prevention of recurrent DVT and PE. The treatment duration was for an additional 6 or 12 months in patients who had completed 6 to 12 months of treatment for venous thromboembolism depending on the clinical judgment of the investigator. Xarelto 20 mg once daily was compared with placebo.
All phase III studies used the same pre-defined primary and secondary efficacy outcomes. The primary efficacy outcome was symptomatic recurrent VTE defined as the composite of recurrent DVT or fatal or non-fatal PE. The secondary efficacy outcome was defined as the composite of recurrent DVT, non-fatal PE and all cause mortality.
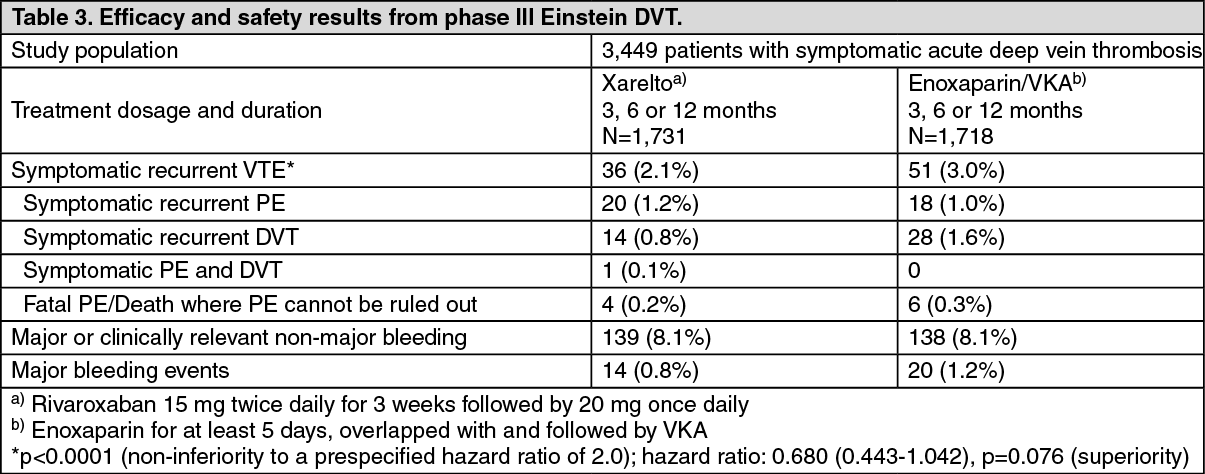
In the Einstein DVT study (see Table 3) rivaroxaban was demonstrated to be non-inferior to enoxaparin/VKA for the primary efficacy outcome (p < 0.0001 (test for non-inferiority); hazard ratio: 0.680 (0.443 - 1.042), p=0.076 (test for superiority)). The prespecified net clinical benefit (primary efficacy outcome plus major bleeding events) was reported with a hazard ratio of 0.67 ((95% CI: 0.47 - 0.95), nominal p value p=0.027) in favour of rivaroxaban. INR values were within the therapeutic range a mean of 60.3% of the time for the mean treatment duration of 189 days, and 55.4%, 60.1%, and 62.8% of the time in the 3-, 6-, and 12-month intended treatment duration groups, respectively. In the enoxaparin/VKA group, there was no clear relation between the level of mean centre TTR (Time in Target INR Range of 2.0 - 3.0) in the equally sized tertiles and the incidence of the recurrent VTE (P=0.932 for interaction). Within the highest tertile according to centre, the hazard ratio with rivaroxaban versus warfarin was 0.69 (95% CI: 0.35 - 1.35).
The incidence rates for the primary safety outcome (major or clinically relevant non-major bleeding events) as well as the secondary safety outcome (major bleeding events) were similar for both treatment groups. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the Einstein PE study (see Table 4) rivaroxaban was demonstrated to be non-inferior to enoxaparin/VKA for the primary efficacy outcome (p=0.0026 (test for non-inferiority); hazard ratio: 1.123 (0.749 - 1.684)). The prespecified net clinical benefit (primary efficacy outcome plus major bleeding events) was reported with a hazard ratio of 0.849 ((95% CI: 0.633 - 1.139), nominal p value p= 0.275). INR values were within the therapeutic range a mean of 63% of the time for the mean treatment duration of 215 days, and 57%, 62%, and 65% of the time in the 3-, 6-, and 12-month intended treatment duration groups, respectively. In the enoxaparin/VKA group, there was no clear relation between the level of mean centre TTR (Time in Target INR Range of 2.0 - 3.0) in the equally sized tertiles and the incidence of the recurrent VTE (p=0.082 for interaction). Within the highest tertile according to centre, the hazard ratio with rivaroxaban versus warfarin was 0.642 (95% CI: 0.277 - 1.484).
The incidence rates for the primary safety outcome (major or clinically relevant non-major bleeding events) were slightly lower in the rivaroxaban treatment group (10.3% (249/2412)) than in the enoxaparin/VKA treatment group (11.4% (274/2405)). The incidence of the secondary safety outcome (major bleeding events) was lower in the rivaroxaban group (1.1% (26/2412)) than in the enoxaparin/VKA group (2.2% (52/2405)) with a hazard ratio 0.493 (95% CI: 0.308 - 0.789). (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA prespecified pooled analysis of the outcome of the Einstein DVT and PE studies was conducted (see Table 5).
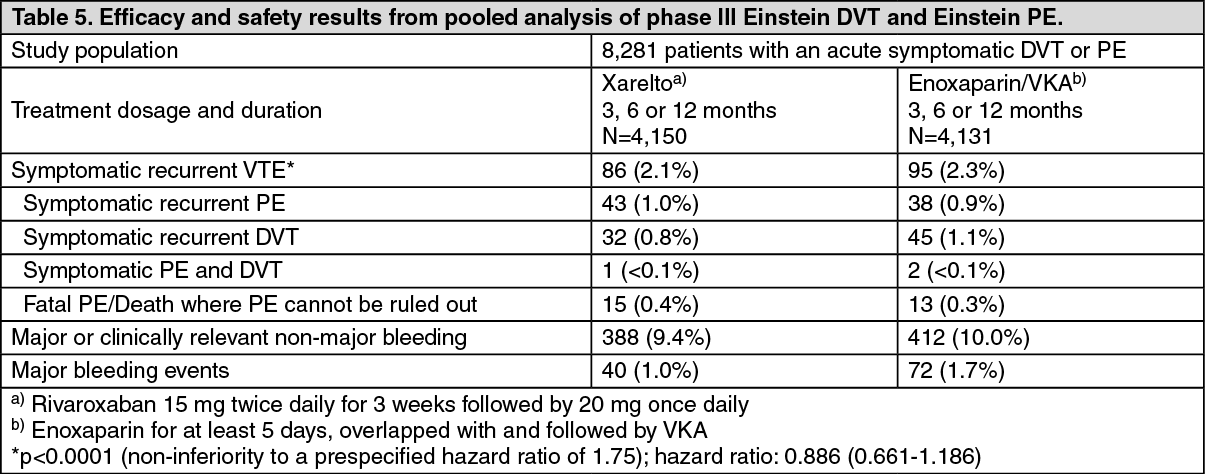 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe prespecified net clinical benefit (primary efficacy outcome plus major bleeding events) of the pooled analysis was reported with a hazard ratio of 0.771 ((95% CI: 0.614 - 0.967), nominal p value p= 0.0244).
In the Einstein Extension study (see Table 6) rivaroxaban was superior to placebo for the primary and secondary efficacy outcomes. For the primary safety outcome (major bleeding events) there was a non-significant numerically higher incidence rate for patients treated with rivaroxaban 20 mg once daily compared to placebo. The secondary safety outcome (major or clinically relevant non-major bleeding events) showed higher rates for patients treated with rivaroxaban 20 mg once daily compared to placebo. (See Table 6.)
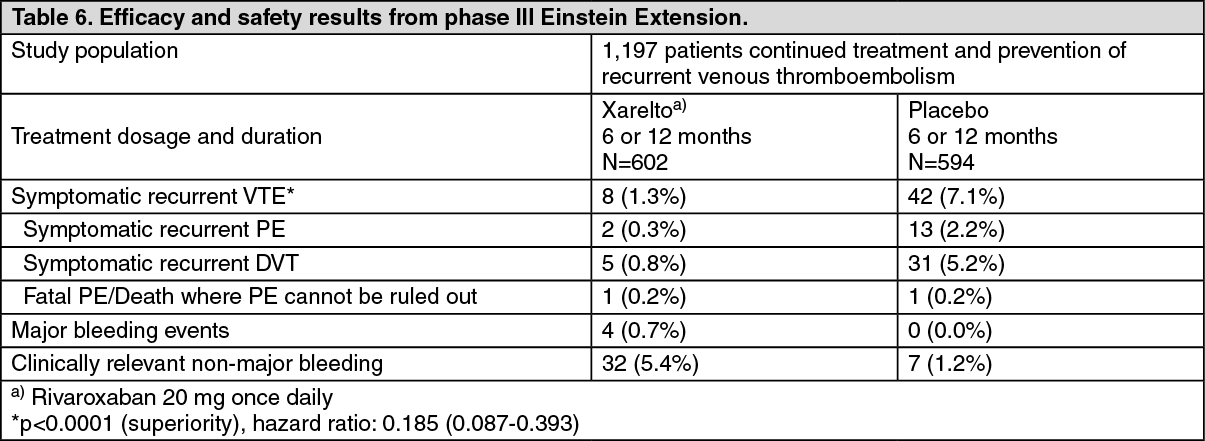 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePaediatric population: The European Medicines Agency has deferred the obligation to submit the results of studies with Xarelto in one or more subsets of the paediatric population in the treatment of thromboembolic events. The European Medicines Agency has waived the obligation to submit the results of studies with Xarelto in all subsets of the paediatric population in the prevention of thromboembolic events (see Dosage & Administration).
Pharmacokinetics: Absorption: Rivaroxaban is rapidly absorbed with maximum concentrations (Cmax) appearing 2 - 4 hours after tablet intake.
Oral absorption of rivaroxaban is almost complete and oral bioavailability is high (80 - 100%) for the 2.5 mg and 10 mg tablet dose, irrespective of fasting/fed conditions. Intake with food does not affect rivaroxaban AUC or Cmax at the 2.5 mg and 10 mg dose.
Due to a reduced extent of absorption an oral bioavailability of 66% was determined for the 20 mg tablet under fasting conditions. When Xarelto 20 mg tablets are taken together with food increases in mean AUC by 39% were observed when compared to tablet intake under fasting conditions, indicating almost complete absorption and high oral bioavailability. Xarelto 15 mg and 20 mg are to be taken with food (see Dosage & Administration).
Rivaroxaban pharmacokinetics are approximately linear up to about 15 mg once daily in fasting state. Under fed conditions Xarelto 10 mg, 15 mg and 20 mg tablets demonstrated dose-proportionality. At higher doses rivaroxaban displays dissolution limited absorption with decreased bioavailability and decreased absorption rate with increased dose.
Variability in rivaroxaban pharmacokinetics is moderate with inter-individual variability (CV%) ranging from 30% to 40%.
Absorption of rivaroxaban is dependent on the site of its release in the gastrointestinal tract. A 29% and 56% decrease in AUC and Cmax compared to tablet was reported when rivaroxaban granulate is released in the proximal small intestine. Exposure is further reduced when rivaroxaban is released in the distal small intestine, or ascending colon. Therefore, administration of rivaroxaban distal to the stomach should be avoided since this can result in reduced absorption and related rivaroxaban exposure.
Bioavailability (AUC and Cmax) was comparable for 20 mg rivaroxaban administered orally as a crushed tablet mixed in apple puree, or suspended in water and administered via a gastric tube followed by a liquid meal, compared to a whole tablet. Given the predictable, dose-proportional pharmacokinetic profile of rivaroxaban, the bioavailability results from this study are likely applicable to lower rivaroxaban doses.
Distribution: Plasma protein binding in humans is high at approximately 92% to 95%, with serum albumin being the main binding component. The volume of distribution is moderate with Vss being approximately 50 litres.
Biotransformation and elimination: Of the administered rivaroxaban dose, approximately 2/3 undergoes metabolic degradation, with half then being eliminated renally and the other half eliminated by the faecal route. The final 1/3 of the administered dose undergoes direct renal excretion as unchanged active substance in the urine, mainly via active renal secretion.
Rivaroxaban is metabolised via CYP3A4, CYP2J2 and CYP-independent mechanisms. Oxidative degradation of the morpholinone moiety and hydrolysis of the amide bonds are the major sites of biotransformation. Based on in vitro investigations rivaroxaban is a substrate of the transporter proteins P-gp (P-glycoprotein) and Bcrp (breast cancer resistance protein).
Unchanged rivaroxaban is the most important compound in human plasma, with no major or active circulating metabolites being present. With a systemic clearance of about 10 l/h, rivaroxaban can be classified as a low-clearance substance. After intravenous administration of a 1 mg dose the elimination half-life is about 4.5 hours. After oral administration the elimination becomes absorption rate limited. Elimination of rivaroxaban from plasma occurs with terminal half-lives of 5 to 9 hours in young individuals, and with terminal half-lives of 11 to 13 hours in the elderly.
Special populations: Gender: There were no clinically relevant differences in pharmacokinetics and pharmacodynamics between male and female patients.
Elderly population: Elderly patients exhibited higher plasma concentrations than younger patients, with mean AUC values being approximately 1.5 fold higher, mainly due to reduced (apparent) total and renal clearance. No dose adjustment is necessary.
Different weight categories: Extremes in body weight (< 50 kg or > 120 kg) had only a small influence on rivaroxaban plasma concentrations (less than 25%). No dose adjustment is necessary.
Inter-ethnic differences: No clinically relevant inter-ethnic differences among Caucasian, African-American, Hispanic, Japanese or Chinese patients were observed regarding rivaroxaban pharmacokinetics and pharmacodynamics.
Hepatic impairment: Cirrhotic patients with mild hepatic impairment (classified as Child Pugh A) exhibited only minor changes in rivaroxaban pharmacokinetics (1.2 fold increase in rivaroxaban AUC on average), nearly comparable to their matched healthy control group. In cirrhotic patients with moderate hepatic impairment (classified as Child Pugh B), rivaroxaban mean AUC was significantly increased by 2.3 fold compared to healthy volunteers. Unbound AUC was increased 2.6 fold. These patients also had reduced renal elimination of rivaroxaban, similar to patients with moderate renal impairment.
There are no data in patients with severe hepatic impairment.
The inhibition of factor Xa activity was increased by a factor of 2.6 in patients with moderate hepatic impairment as compared to healthy volunteers; prolongation of PT was similarly increased by a factor of 2.1. Patients with moderate hepatic impairment were more sensitive to rivaroxaban resulting in a steeper PK/PD relationship between concentration and PT.
Xarelto is contraindicated in patients with hepatic disease associated with coagulopathy and clinically relevant bleeding risk, including cirrhotic patients with Child Pugh B and C (see Contraindications).
Renal impairment: There was an increase in rivaroxaban exposure correlated to decrease in renal function, as assessed via creatinine clearance measurements. In individuals with mild (creatinine clearance 50 - 80 ml/min), moderate (creatinine clearance 30 - 49 ml/min) and severe (creatinine clearance 15 - 29 ml/min) renal impairment, rivaroxaban plasma concentrations (AUC) were increased 1.4, 1.5 and 1.6 fold respectively. Corresponding increases in pharmacodynamic effects were more pronounced. In individuals with mild, moderate and severe renal impairment the overall inhibition of factor Xa activity was increased by a factor of 1.5, 1.9 and 2.0 respectively as compared to healthy volunteers; prolongation of PT was similarly increased by a factor of 1.3, 2.2 and 2.4 respectively. There are no data in patients with creatinine clearance < 15 ml/min.
Due to the high plasma protein binding rivaroxaban is not expected to be dialysable.
Use is not recommended in patients with creatinine clearance < 15 ml/min. Xarelto is to be used with caution in patients with creatinine clearance 15 - 29 ml/min (see Precautions).
Pharmacokinetic data in patients: In patients receiving rivaroxaban for treatment of acute DVT 20 mg once daily the geometric mean concentration (90% prediction interval) 2 - 4 h and about 24 h after dose (roughly representing maximum and minimum concentrations during the dose interval) was 215 (22 - 535) and 32 (6 - 239) μg/l, respectively.
Pharmacokinetic/pharmacodynamic relationship: The pharmacokinetic/pharmacodynamic (PK/PD) relationship between rivaroxaban plasma concentration and several PD endpoints (factor Xa inhibition, PT, aPTT, Heptest) has been evaluated after administration of a wide range of doses (5 - 30 mg twice a day). The relationship between rivaroxaban concentration and factor Xa activity was best described by an Emax model. For PT, the linear intercept model generally described the data better. Depending on the different PT reagents used, the slope differed considerably. When Neoplastin PT was used, baseline PT was about 13 s and the slope was around 3 to 4 s/(100 µg/l). The results of the PK/PD analyses in Phase II and III were consistent with the data established in healthy subjects.
Paediatric population: Safety and efficacy have not been established for children and adolescents up to 18 years.
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, single dose toxicity, phototoxicity, genotoxicity, carcinogenic potential and juvenile toxicity.
Effects observed in repeat-dose toxicity studies were mainly due to the exaggerated pharmacodynamic activity of rivaroxaban. In rats, increased IgG and IgA plasma levels were seen at clinically relevant exposure levels.
In rats, no effects on male or female fertility were seen. Animal studies have shown reproductive toxicity related to the pharmacological mode of action of rivaroxaban (e.g. haemorrhagic complications). Embryo-foetal toxicity (post-implantation loss, retarded/progressed ossification, hepatic multiple light coloured spots) and an increased incidence of common malformations as well as placental changes were observed at clinically relevant plasma concentrations. In the pre- and postnatal study in rats, reduced viability of the offspring was observed at doses that were toxic to the dams.