Thrombopoietin-receptor agonist.
Pharmacology: Mechanism of action (MOA): Thrombopoietin (TPO) is the main cytokine involved in regulation of megakaryopoiesis and platelet production, and is the endogenous ligand for the TPO-receptor. Eltrombopag interacts with the transmembrane domain of the human TPO-receptor and initiates signaling cascades similar but not identical to that of endogenous TPO, inducing proliferation and differentiation of megakaryocytes and bone marrow progenitor cells.
Pharmacodynamics (PD): Eltrombopag differs from TPO with respect to the effects on platelet aggregation. Unlike TPO, eltrombopag treatment of normal human platelets does not enhance adenosine diphosphate (ADP)-induced aggregation or induce P-selectin expression. Eltrombopag does not antagonize platelet aggregation induced by ADP or collagen.
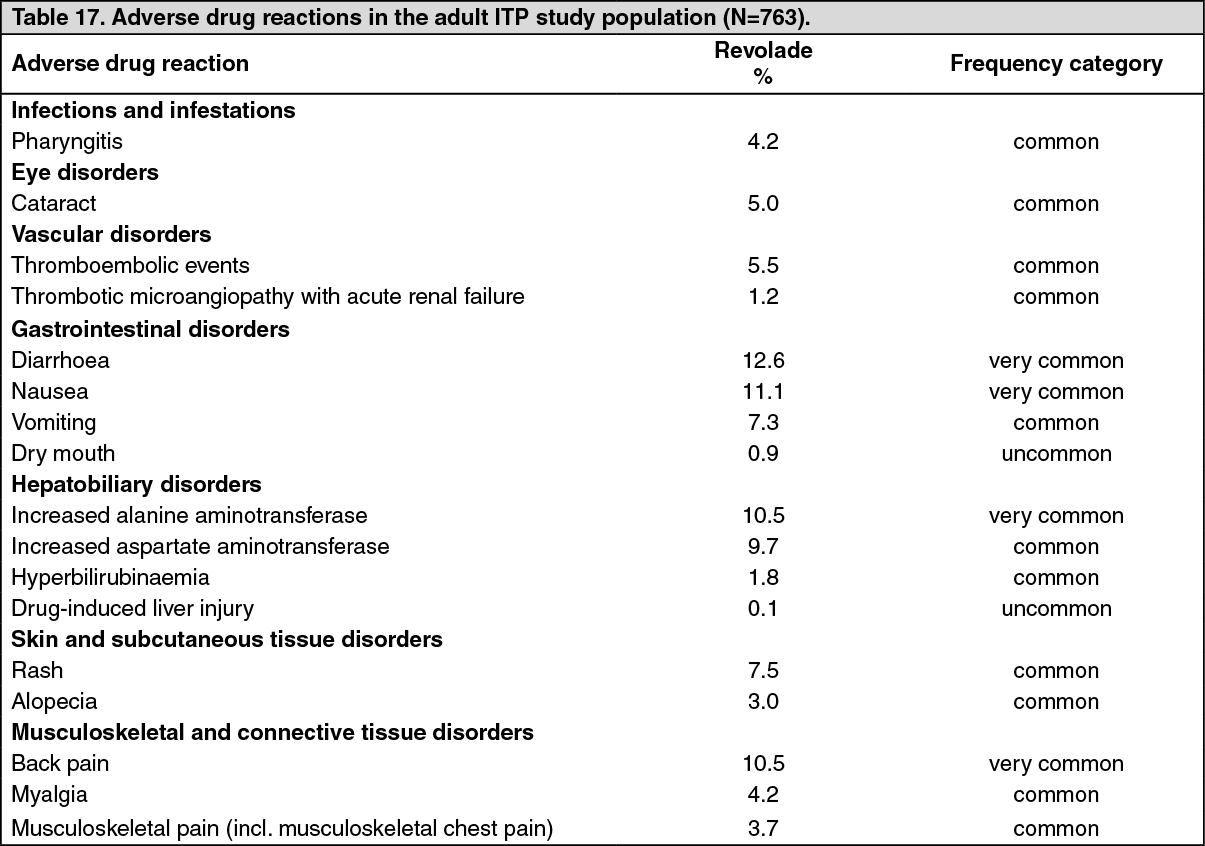
Clinical Studies: Immune thrombocytopenia (ITP) studies: Adults: The safety and efficacy of Revolade in adult patients with previously treated ITP have been demonstrated in two, randomized, double-blind, placebo-controlled studies (TRA102537 [RAISE] and TRA100773B) and two open label studies (TRA108057 [REPEAT] and TRA105325 [EXTEND]).
Double-blind placebo-controlled studies: TRA102537 (RAISE): In TRA102537, the primary efficacy endpoint was the odds of achieving a platelet count ≥50,000/microL and ≤400,000/microL, during the 6 month treatment period, for patients receiving Revolade relative to placebo. One hundred and ninety-seven patients were randomized 2:1 to Revolade (n = 135) and to placebo (n = 62). Stratification was based upon splenectomy status, use of ITP medication at baseline, and baseline platelet count. Patients received study medication for up to 6 months, during which time the dose of Revolade could be adjusted based on individual platelet counts. In addition, patients could have tapered off concomitant ITP medications and received rescue treatments according to local standard of care.
The odds of achieving a platelet count between 50,000/microL and 400,000/microL during the 6-month treatment period were 8 times higher for Revolade treated patients than for placebo-treated patients (Odds Ratio (OR): 8.2 [99% CI: 3.59, 18.73] p = <0.001). Median platelet counts were maintained above 50,000/microL at all on-therapy visits starting at Day-15 in the Revolade group; in contrast, median platelet counts in the placebo group remained below 30,000/microL throughout the study.
At baseline, 77% of patients in the placebo group and 73% of patients in the Revolade group reported any bleeding (WHO Grades 1-4); clinically significant bleeding (WHO Grades 2-4) at baseline was reported in 28% and 22% of patients in the placebo and Revolade groups, respectively. The proportion of patients with any bleeding (Grades 1-4) and clinically significant bleeding (Grades 2-4) was reduced from baseline by approximately 50% throughout the 6 month treatment period in Revolade treated patients. When compared to the placebo group, the odds of any bleeding (Grades 1-4) and the odds of clinically significant bleeding (Grades 2-4) were 76% and 65% lower in the Revolade-treated patients compared to the placebo-treated patients (p < 0.001).
Revolade therapy allowed significantly more patients to reduce or discontinue baseline ITP therapies compared to placebo (59% vs. 32%; p <0.016).
Significantly fewer Revolade-treated patients required rescue treatment compared to placebo-treated patients (18% vs. 40%; p = 0.001).
Four placebo and 14 Revolade patients had at least 1 hemostatic challenge (defined as an invasive diagnostic or surgical procedure) during the study. Fewer Revolade-treated patients (29%) required rescue treatment to manage their hemostatic challenge, compared to placebo-treated patients (50%).
In terms of improvements in health-related quality of life, statistically significant improvements from baseline were observed in the Revolade group in fatigue, including severity and impact on thrombocytopenia-impacted daily activities and concerns (as measured by the vitality subscale of the SF36, the motivation and energy inventory, and the 6-item extract from the thrombocytopenia subscale of the FACIT-Th). Comparing the Revolade group to the placebo group, statistically significant improvements were observed with thrombocytopenia impacted activities and concerns specifically regarding motivation, energy and fatigue, as well as physical and emotional role and overall mental health. The odds of meaningful improvement in health related quality of life while on therapy were significantly greater among patients treated with Revolade than placebo.
TRA100773B: In TRA100773B, the primary efficacy endpoint was the proportion of responders, defined as patients who had an increase in platelet counts to ≥50,000/microL at Day 43 from a baseline platelet count <30,000/microL; patients who withdrew prematurely due to a platelet count >200,000/microL were considered responders, those discontinued for any other reason were considered non-responders irrespective of platelet count. A total of 114 patients with previously treated ITP were randomized 2:1, with 76 randomized to Revolade and 38 randomized to placebo.
Fifty-nine percent of patients on Revolade responded, compared to 16% of patients on placebo. The odds of responding were 9 times higher for Revolade treated patients compared to placebo (OR: 9.6 [95% CI: 3.31, 27.86] p <0.001). At baseline, 61% of patients in the Revolade group and 66% of patients in the placebo group reported any bleeding (Grade 1-4). At Day 43, 39% of patients in the Revolade treatment group had bleeding compared with 60% in the placebo group. Analysis over the treatment period using a repeated measures model for binary data confirmed that a lower proportion of Revolade patients had bleeding (Grade 1-4) at any point in time over the course of their treatment (Day 8 up to Day 43) compared to patients in the placebo group (OR:0.49, (95% CI: 0.26, 0.89), p = 0.021). Two placebo and one Revolade patient had at least one hemostatic challenge during the study.
In both RAISE and TRA100773B the response to Revolade relative to placebo was similar irrespective of ITP medication use, splenectomy status and baseline platelet count (≤15,000/microL, >15,000/microL) at randomization.
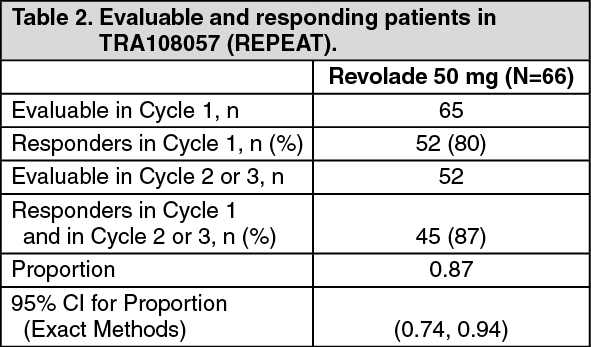
Open label studies: TRA108057 (REPEAT): TRA108057 was an open-label, repeat-dose study which evaluated the efficacy, safety and consistency of response following repeated, intermittent, short-term dosing of Revolade over 3 cycles of therapy in adults with previously treated ITP. A cycle was defined as an up to 6-week on-therapy period followed by an up to 4-week off-therapy period. The duration of both the on-therapy and the off-therapy periods was defined by the patient's platelet count. Patients were to interrupt treatment for the cycle if they achieved a platelet count >200,000/microL, or when they reached Week 6. Patients were to begin the next cycle when their platelet counts fell below 20,000/microL, or when they reached Week 4 of the off-therapy period. The primary endpoint was the proportion of patients who achieved a platelet count ≥ 50,000/microL and at least 2 x baseline in Cycle 2 or 3, given this response in Cycle 1. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Of the 52 patients who responded in Cycle 1, 33 (63%) achieved a platelet count of ≥50,000 microL and at least 2 x baseline on Day 8 in Cycle 1; on Day 15, 37 (79%) of 47 evaluable patients achieved this level of response.
A reduction in any bleeding (WHO Grade 1-4) and clinically significant bleeding (WHO Grade 2-4) during the treatment phases was demonstrated in each cycle. At the baseline visit of Cycle 1, 50% and 19% of patients reported any bleeding and clinically significant bleeding, respectively. At the Day 43 Visit of Cycle 1, the proportion of patients bleeding was reduced; 12% and 0% of patients reported any bleeding and clinically significant bleeding. Similar results were found during the subsequent treatment cycles.
Eight patients successfully managed 10 hemostatic challenges without need for additional therapy to elevate platelet counts and without unexpected bleeding.
TRA105325 (EXTEND): TRA105325 was an open label extension study which has evaluated the safety and efficacy of Revolade in patients with ITP at least 6 months from diagnosis who were previously enrolled in a Revolade study. In this study, patients were permitted to modify their dose of study medication as well as decrease or eliminate concomitant ITP medications.
Revolade was administered to 302 ITP patients; 218 completed 1 year of treatment, 180 completed 2 years, 107 completed 3 years, 75 completed 4 years, 34 completed 5 years and 18 completed 6 years of therapy. The median baseline platelet count was 19,000/microL prior to Revolade administration. Median platelet counts at 1, 2, 3, 4, 5, 6 and 7 years on study were 85,000/microL, 85,000/microL, 105,000/microL, 64,000/microL, 75,000/microL, 119,000/microL and 76,000/microL respectively. The median daily dose of Revolade following 6 months of therapy was 50 mg (n = 74).
At baseline, 59% of patients had any bleeding (WHO Bleeding Grades 1-4) and 18% had clinically significant bleeding (WHO Bleeding Grades 2 indicating clinically significant bleeding). The proportion of patients with any bleeding and clinically significant bleeding decreased from baseline by approximately 50% for the majority of assessments up to 1 year.
One-hundred and one patients were taking ITP medications at baseline upon entry into EXTEND study, and 39 patients were able to permanently discontinue or achieve a sustained reduction of at least one baseline ITP medication without needing rescue medication. Sixty-five percent of these patients maintained this discontinuation or reduction for at least 24 weeks. Sixty-one percent of patients completely discontinued at least one baseline ITP medication, and 55% of patients permanently discontinued all baseline ITP medications, without subsequent rescue treatment.
Twenty-four patients experienced at least one hemostatic challenge during the study. No patient experienced unexpected bleeding complications related to the procedure while on study.
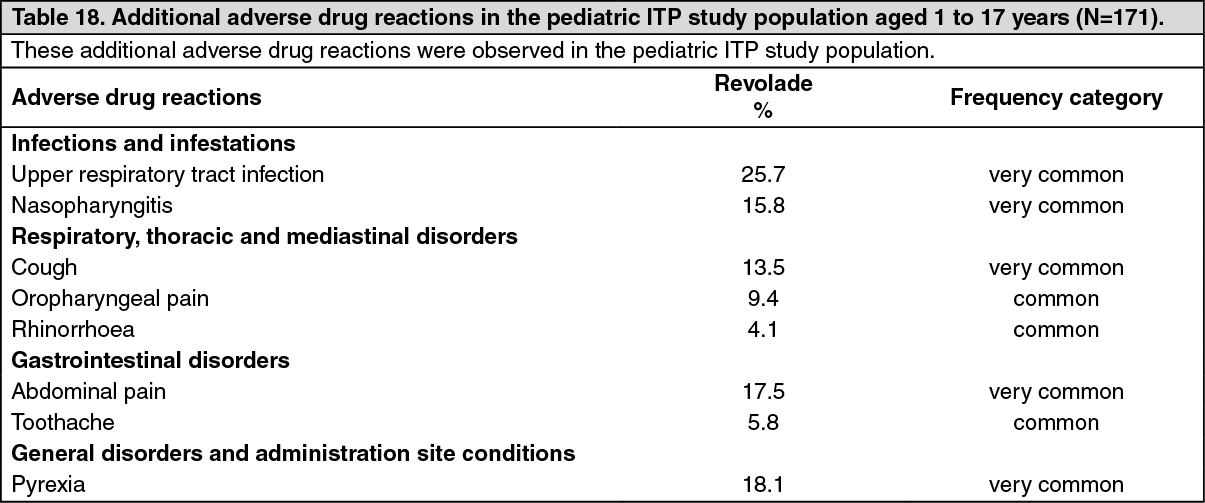
Pediatric patients (aged 1 to 17 years): The safety and efficacy of Revolade in pediatric patients with previously treated ITP have been demonstrated in two studies.
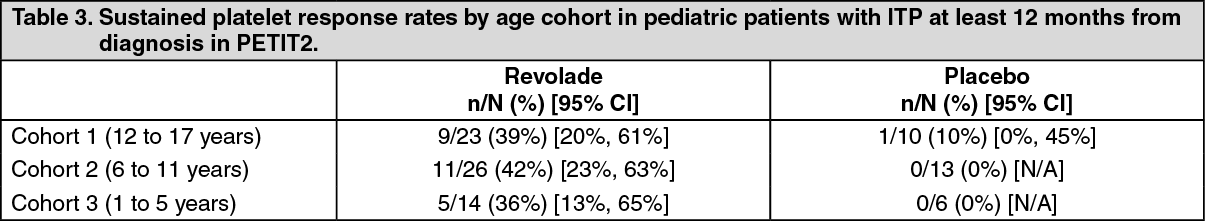
Double-blind placebo-controlled studies: TRA115450 (PETIT2): The primary endpoint was a sustained response, defined as the proportion of patients receiving Revolade, compared to placebo, achieving platelet counts ≥50,000/microL for at least 6 out of 8 weeks (in the absence of rescue therapy), between Weeks 5 to 12 during the double-blind randomized period. Patients were refractory or relapsed to at least one prior ITP therapy or unable to continue other ITP treatments for a medical reason and had platelet count <30,000/microL. Ninety-two patients were randomized by three age cohort strata (2:1) to Revolade (n = 63) or placebo (n = 29). The dose of Revolade could be adjusted based on individual platelet counts.
Overall, a significantly greater proportion of Revolade patients (40%) compared with placebo patients (3%) achieved the primary endpoint (OR: 18.0 (95% CI: 2.3, 140.9) p< 0.001) which was similar across the three age cohorts (Table 3). (See Table 3.)
Click on icon to see table/diagram/image
A significantly greater proportion of patients treated with Revolade (75%) compared with placebo (21%) had a platelet response (at least one platelet count >50,000/microL during the first 12 weeks of randomized treatment in absence of rescue therapy) (OR: 11.7, (95% CI: 4.0, 34.5), p <0.001). The proportion of patients who responded to Revolade in the open-label 24-week period (80%) was similar to that observed during the randomized portion of the study.
Statistically fewer Revolade patients required rescue treatment during the randomized period compared to placebo patients (19% (12/63) vs. 24% (7/29), p = 0.032).
At baseline, 71% of patients in the Revolade group and 69% in the placebo group reported any bleeding (WHO Grades 1-4). At Week 12, the proportion of Revolade patients reporting any bleeding was decreased to half of baseline (36%). In comparison, at Week 12, 55% of placebo patients reported any bleeding.
Patients were permitted to reduce or discontinue baseline ITP therapy only during the open-label phase of the study and 53% (8/15) of patients were able to reduce (n = 1) or discontinue (n = 7) baseline ITP therapy, mainly corticosteroids, without needing rescue therapy.
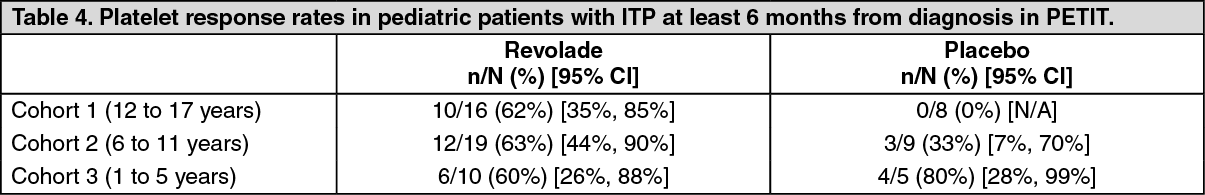
TRA108062 (PETIT): The primary endpoint was the proportion of patients achieving platelet counts ≥50,000/ microL at least once between Weeks 1 and 6 of the randomized period. Patients were refractory or relapsed to at least one prior ITP therapy with a platelet count <30,000/ (n = 67). During the randomized period of the study, patients were randomized by 3 age cohort strata (2:1) to Revolade (n = 45) or placebo (n = 22). The dose of Revolade could be adjusted based on individual platelet counts.
Overall, a significantly greater proportion of Revolade patients (62%) compared with placebo patients (32%) met the primary endpoint (OR: 4.3 (95% CI: 1.4, 13.3) p = 0.011). Table 4 shows platelet response across the three age cohorts. (See Table 4.)
Click on icon to see table/diagram/image
A significantly greater proportion of patients treated with Revolade (36%) compared with placebo (0%) had a platelet response (platelet counts >50,000/microL for at least 60% of assessments between Weeks 2 and 6) (OR: 5.8, (95% CI: 1.2, 28.9), p = 0.002).
Statistically fewer Revolade-treated patients required rescue treatment during the randomized period compared to placebo treated patients (13% (6/45) vs. 50% (11/22), p = 0.002).
At baseline, 77.7% of patients in the Revolade group and 81.8% in the placebo group reported any bleeding (WHO Grades 1-4). The proportion of Revolade patients reporting any bleeding decreased to 22.2% at Week 6. In comparison, 72.7% of placebo patients reported any bleeding at Week 6.
Patients were permitted to reduce or discontinue baseline ITP therapy only during the open-label phase of the study and 46% (6/13) of patients were able to reduce (n = 3) or discontinue (n = 3) baseline ITP therapy, mainly corticosteroids, without needing rescue therapy.
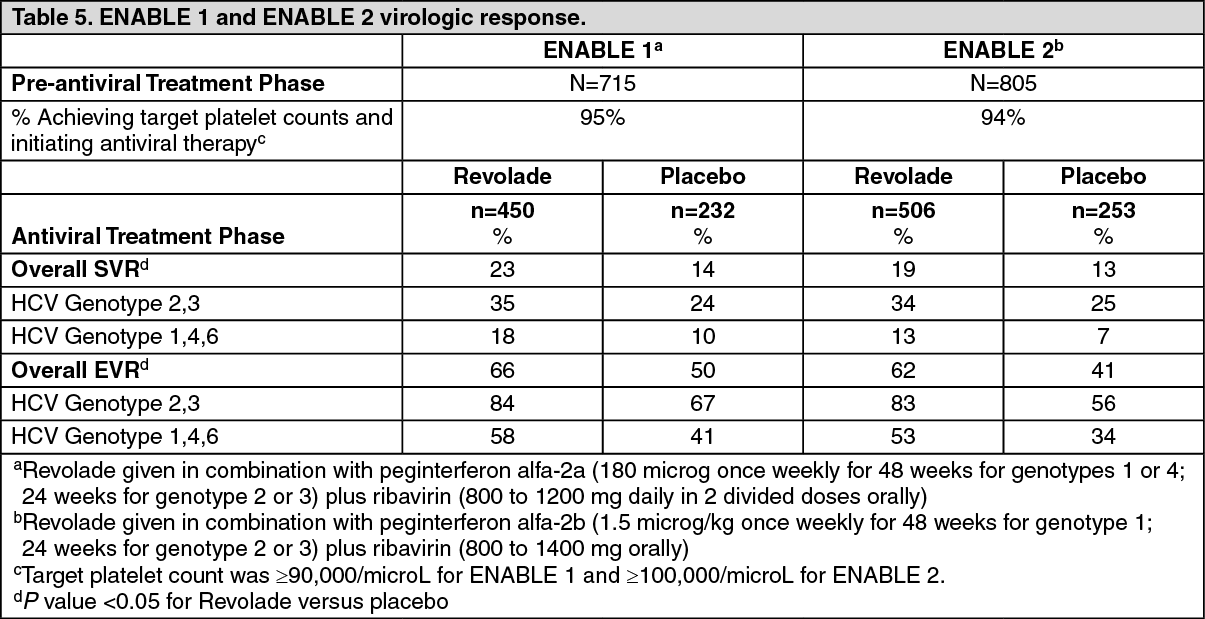
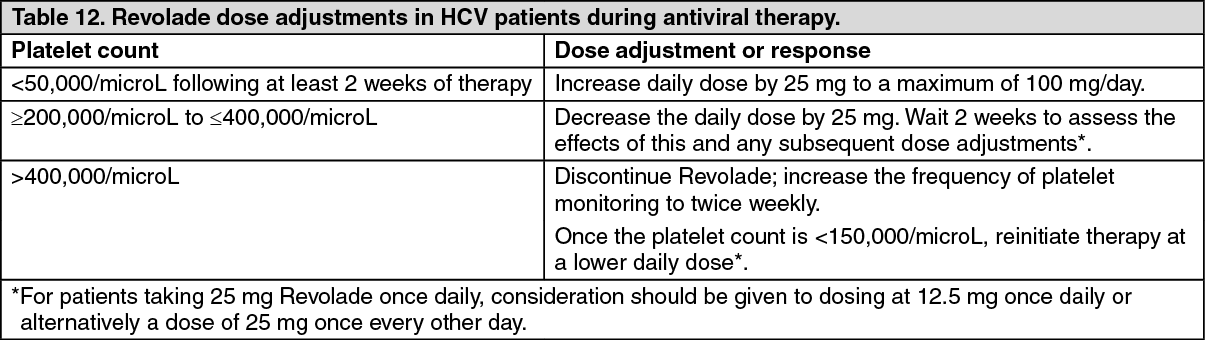
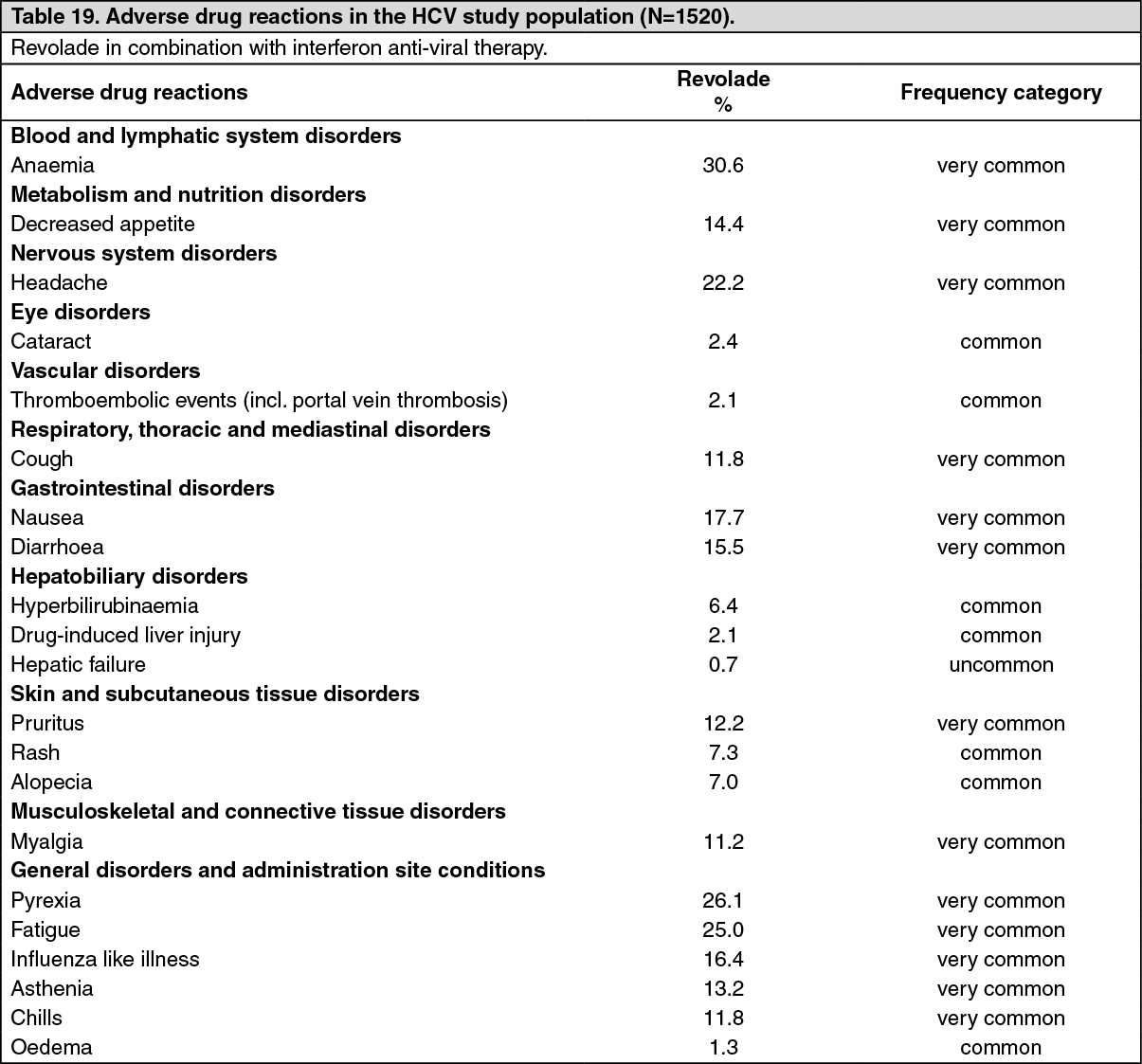
Chronic hepatitis C associated thrombocytopenia studies: The efficacy and safety of Revolade for the treatment of thrombocytopenia in patients with HCV infection were evaluated in two randomized, double-blind, placebo-controlled studies. ENABLE 1 utilized peginterferon alfa-2a plus ribavirin for antiviral treatment and ENABLE 2 utilized peginterferon alfa-2b plus ribavirin. In both studies, patients with a platelet count of <75,000/microL were enrolled and stratified by platelet count (<50,000/microL and ≥50,000/microL to <75,000/microL), screening HCV RNA (<800,000 IU/mL and ≥800,000 IU/mL), and HCV genotype (genotype 2/3, and genotype 1/4/6).
The studies consisted of two phases-a pre-antiviral treatment phase and an antiviral treatment phase. In the pre-antiviral treatment phase, patients received open-label Revolade to increase the platelet count to ≥90,000/microL for ENABLE 1 and ≥100,000/microL for ENABLE 2. Revolade was administered at an initial dose of 25 mg once daily for 2 weeks and increased in 25 mg increments over 2 to 3 week periods to achieve the required platelet count for phase 2 of the study. The maximal time patients could receive open-label Revolade was 9 weeks. If sufficient platelet counts were achieved, patients were randomized (2:1) to the same dose of Revolade at the end of the pre-treatment phase or to placebo. Revolade was administered in combination with antiviral treatment per their respective prescribing information for up to 48 weeks.
The primary efficacy endpoint for both studies was sustained virological response (SVR), defined as the percentage of patients with no detectable HCV-RNA at 24 weeks after completion of the planned treatment period. Approximately 70% of patients were genotype 1/4/6 and 30% were genotype 2/3. Approximately 31% of patients had been treated with prior HCV therapies, primarily pegylated interferon plus ribavirin. The median baseline platelet counts (approximately 60,000/microL) were similar among all treatment groups. The median time to achieve the target platelet count ≥ 90,000/microL (ENABLE 1) or ≥100,000/microL (ENABLE 2) was 2 weeks.
In both HCV studies, a significantly greater proportion of patients treated with Revolade achieved SVR compared to those treated with placebo (see Table 5). Significantly fewer patients treated with Revolade had any antiviral dose reductions compared to placebo. The proportion of patients with no antiviral dose reductions was 45% for Revolade compared to 27% for placebo. Significantly fewer patients treated with Revolade prematurely discontinued antiviral therapy compared to placebo (45% vs. 60%, p = <0.0001). The majority of patients treated with Revolade (76%) had minimum platelet counts that were ≥50,000/microL compared to 19% for placebo. A greater proportion of patients in the placebo group (20%) had minimum platelet counts fall below 25,000/microL during treatment compared to the Revolade group (3%). In the Revolade group, SVR rates in patients with high viral loads (>800,000) were 18% as compared to 8% in the placebo group. Significantly more patients reached the later antiviral milestones of early virologic response (EVR), complete early virologic response (cEVR), end of treatment response (ETR) and sustained virologic response at 12-week follow-up (SVR12) when treated with Revolade. (See Table 5.)
Click on icon to see table/diagram/image
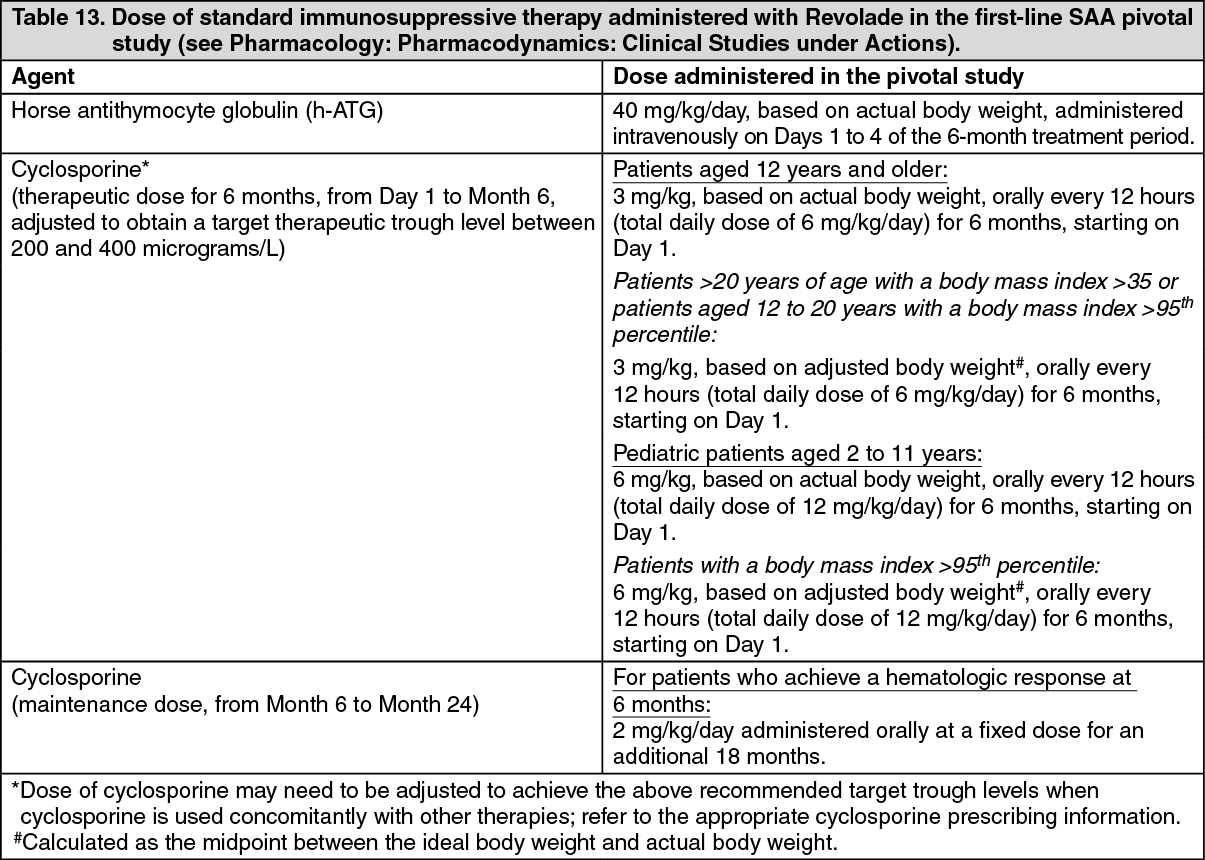
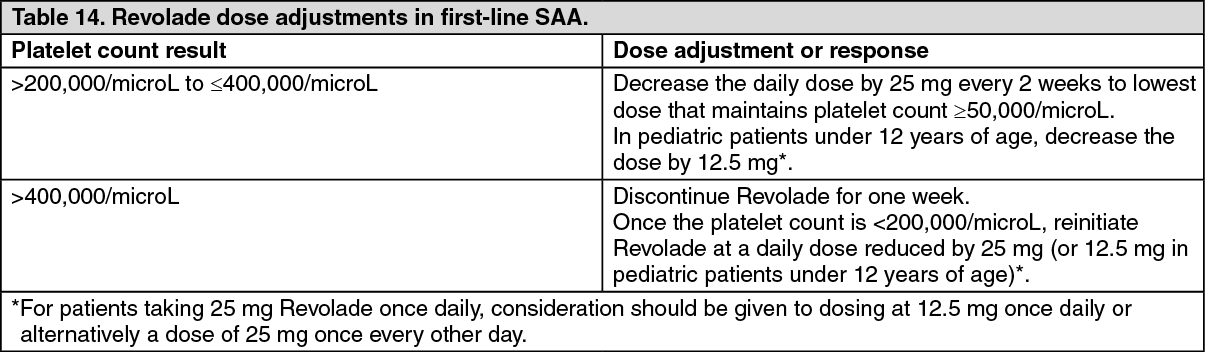
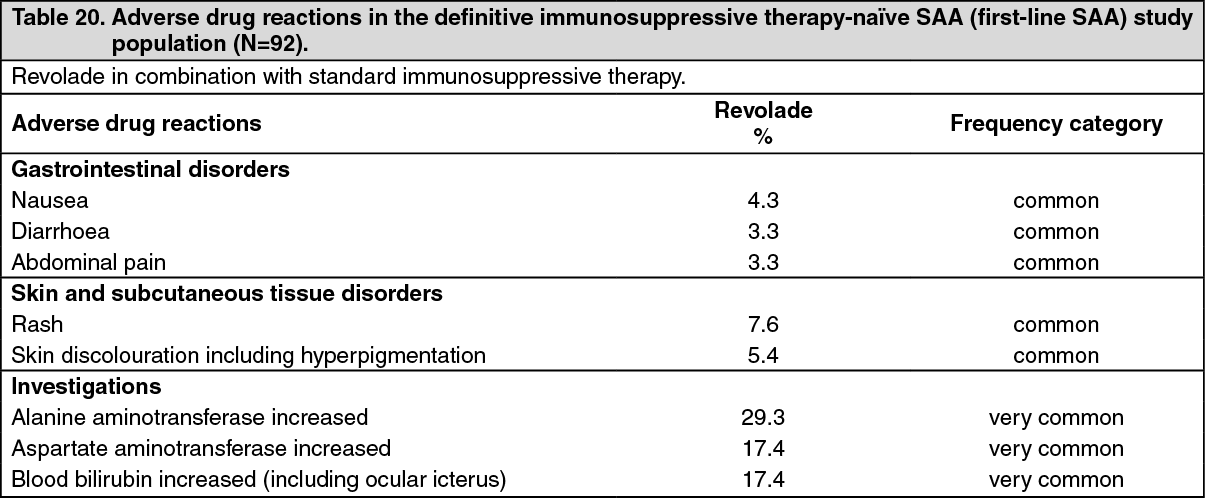
Definitive immunosuppressive therapy-naive severe aplastic anemia study: CETB115AUS01T: Revolade in combination with horse antithymocyte globulin (h-ATG) and cyclosporine was investigated in a single-arm, single-center, open-label, sequential cohort study in patients with severe aplastic anemia who had not received prior definitive immunosuppressive therapy (i.e., ATG therapy, alemtuzumab, or high dose cyclophosphamide). The multiple cohorts differed by treatment start day and duration of Revolade-treatment and the initiation of low dose of cyclosporine (maintenance dose) for patients who achieved a hematologic response at 6 months. A total of 153 patients received Revolade in sequential cohorts: Revolade on Day 14 to Month 6 (D14-M6) plus h-ATG and cyclosporine (Cohort 1 regimen, n=30).
Revolade on Day 14 to Month 3 (D14-M3) plus h-ATG and cyclosporine (Cohort 2 regimen, n=31), with half of the patients eligible to receive low dose of cyclosporine (maintenance dose) if they achieved a hematologic response at 6 months.
Revolade on Day 1 to Month 6 (D1-M6) plus h-ATG and cyclosporine (Cohort 3 regimen, n=92), with all patients eligible to receive low dose of cyclosporine (maintenance dose) if they achieved a hematologic response at 6 months.
The starting dose of Revolade for adults and pediatric patients aged 12 to 17 years was 150 mg once daily (a reduced dose of 75 mg was administered for East-/Southeast-Asians), 75 mg once daily for patients aged 6 to 11 years (a reduced dose of 37.5 mg was administered for East- /Southeast-Asians), and 2.5 mg/kg once daily for patients aged 2 to 5 years (a reduced dose of 1.25 mg/kg was administered for East-/Southeast-Asians). The dose of Revolade was reduced if the platelet count exceeded 200,000/microL and interrupted and reduced if it exceeded 400,000/microL.
All patients received h-ATG 40 mg/kg/day on Days 1 to 4 of the 6-month treatment period and a total daily dose of 6 mg/kg/day of cyclosporine for 6 months in patients aged 12 years and older or a total daily dose of 12 mg/kg/day for 6 months in patients aged 2 to 11 years [61]. A 2 mg/kg/day maintenance dose of cyclosporine was administered for an additional 18 months to 15 patients who achieved a hematologic response at 6 months in the Revolade D14-M3 cohort and all patients who achieved a hematologic response at 6 months in the Revolade D1-M6 cohort.
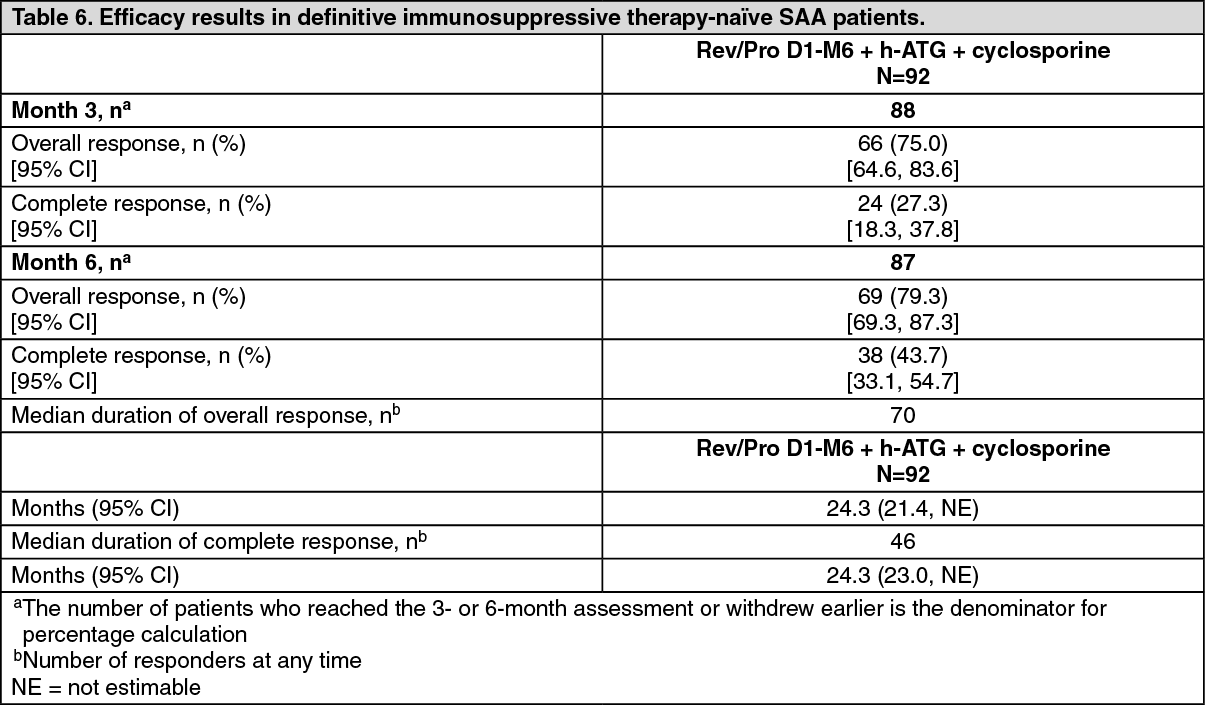
Data from the recommended schedule of Revolade on Day 1 to Month 6 in combination with h-ATG and cyclosporine (Cohort 3 regimen) are presented as follows. This cohort had the highest complete response rates.
In the Revolade D1-M6 cohort, the median age was 28.0 years (range 5 to 82 years) with 16.3% and 28.3% of patients ≥65 years of age and <18 years of age, respectively. 45.7% of patients were male and the majority of patients were White (62.0%).
The efficacy of Revolade in combination with h-ATG and cyclosporine was established on the basis of complete hematological response at 6 months. A complete response was defined as hematological parameters meeting all 3 of the following values on 2 consecutive serial blood count measurements at least one week apart: absolute neutrophil count (ANC) >1,000/microL, platelet count >100,000/microL and hemoglobin >10 g/dL. A partial response was defined as blood counts no longer meeting the standard criteria for severe pancytopenia in severe aplastic anemia equivalent to 2 of the following values on 2 consecutive serial blood count measurements at least one week apart: ANC >500/microL, platelet count >20,000/microL, or reticulocyte count >60,000/microL. (See Table 6.)
Click on icon to see table/diagram/image
The overall and complete hematological response rates at Year 1 (N=78) are 56.4% and 38.5% and at Year 2 (N=62) are 38.7% and 30.6%, respectively.
Pediatric patients: Thirty-seven patients aged 2 to 17 years were enrolled in the single-arm, sequential-cohort study. Of the 36 patients who reached the 6-month assessment point or withdrew earlier, the complete response rate at 6 months was 30.6% (0/2 in patients aged 2 to 5 years, 1/12 in patients aged 6 to 11 years, and 10/22 in patients aged 12 to 17 years) and the overall response rate at 6 months was 72.2% (2/2 in patients aged 2 to 5 years, 7/12 in patients aged 6 to 11 years, and 17/22 in patients aged 12 to 17 years). Out of 25 evaluable patients in the Revolade D1-M6 cohort, the complete response rate at 6 months was 28% (7/25) and the overall response rate at 6 months was 68.0%.
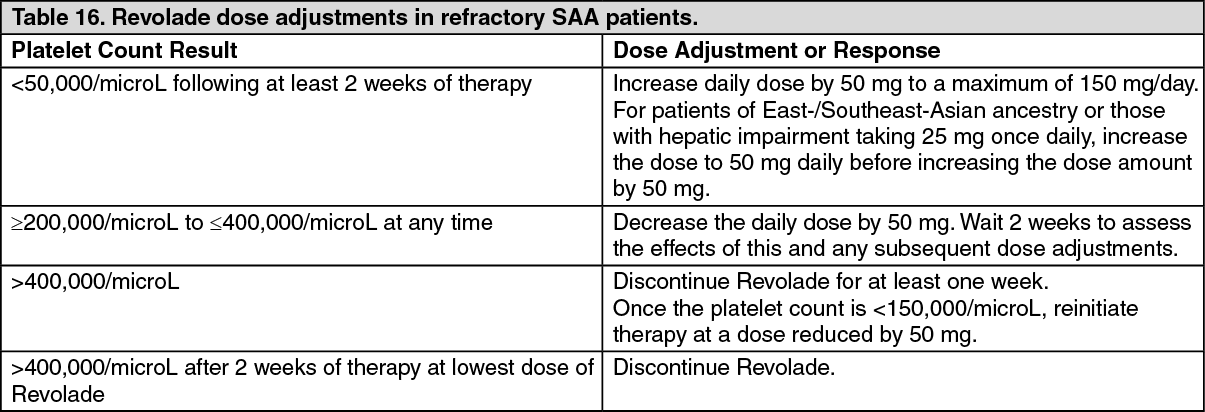
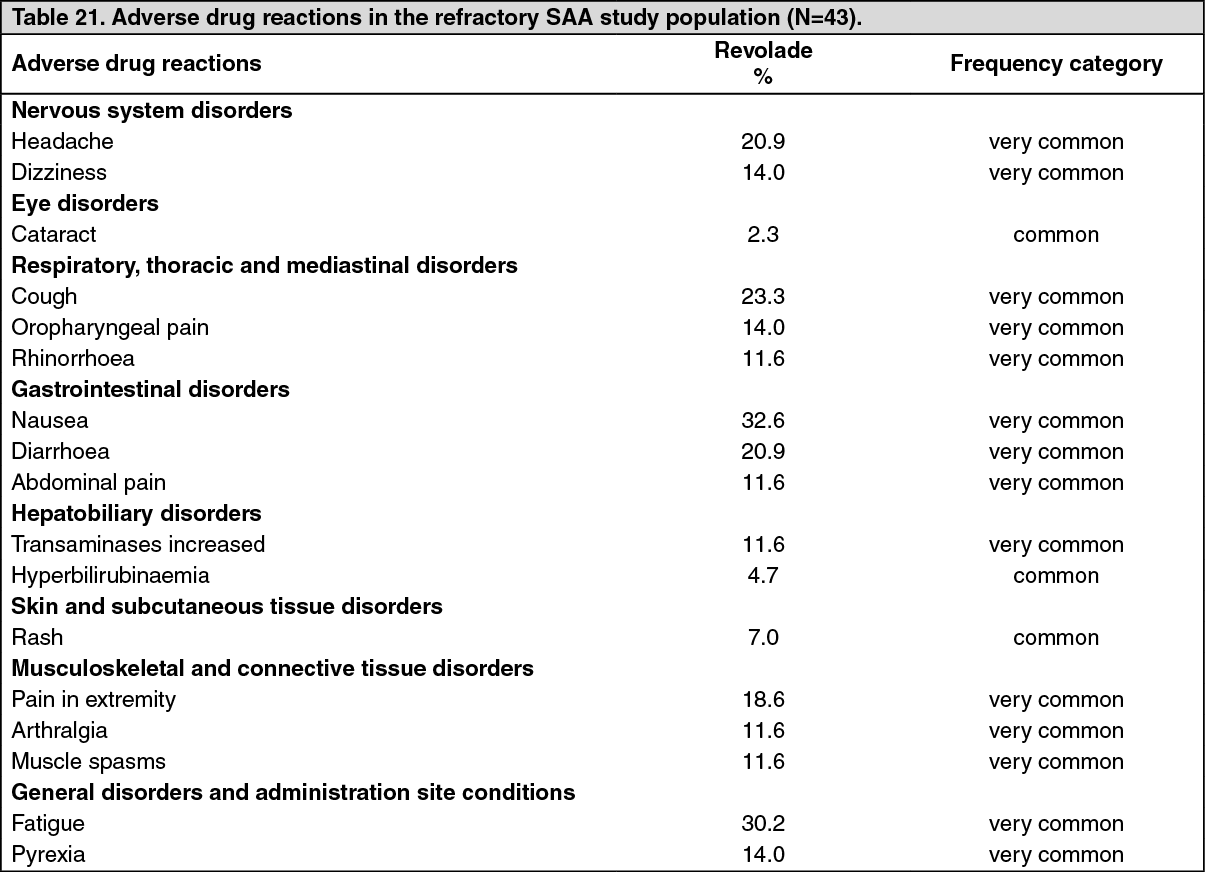
Refractory severe aplastic anemia study: CETB115AUS28T: Revolade was studied in a single-arm, single-center open-label study in 43 patients with severe aplastic anemia who had an insufficient response to at least one prior immunosuppressive therapy and who had a platelet count ≤30,000/microL.
Revolade was administered at an initial dose of 50 mg once daily for 2 weeks and increased over 2 week periods up to a maximum dose of 150 mg once daily. The primary endpoint was hematological response assessed after 12 weeks of Revolade treatment.
Revolade was discontinued after 16 weeks if no hematological response or transfusion independence was observed. Patients who responded continued therapy in an extension phase of the study.
Hematological response was defined as meeting one or more of the following criteria: 1) platelet count increases to 20,000/microL above baseline or stable platelet counts with transfusion independence for a minimum of 8 weeks; 2) hemoglobin increase by >1.5 g/dL (for patients with pre-treatment hemoglobin <9 g/dL), or a reduction in the volume of RBC transfusions of at least 4 units for 8 consecutive weeks; 3) absolute neutrophil count (ANC) increase of 100% (for patients with pre-treatment ANC <500/microL) or an ANC increase 500/microL.
The treated population had median age of 45 years (range 17 to 77 years) and 56% of patients were male. At baseline the median platelet count was 20,000/microL hemoglobin was 8.4 g/dL, and ANC was 580/microL. Eighty-six percent of patients were RBC transfusion dependent, and 91% were platelet transfusion dependent. The majority of patients (84%) had received at least 2 prior immunosuppressive therapies. Three patients had cytogenetic abnormalities at baseline.
A total of 17 patients (40%) met the hematologic response criteria in at least 1 lineage at the Primary Response Assessment (95% CI: 25, 56).
Multi-lineage responses were observed in 4/17 responders (24%) at the initial response assessment and in 9/17 responders (53%) at last assessment. Of the five patients who met protocol specified 'tri-lineage hematopoiesis' criteria for at least eight weeks and were tapered off Revolade, all five patients have maintained tri-lineage hematopoiesis since discontinuing treatment for a median follow up period of 20.6 months (range 5.7 to 22.5 months).
The majority of responders met platelet response criteria (65%), followed by neutrophil and hemoglobin response criteria (47% and 18% respectively). The 15 responders who had at least 2 response assessments were evaluable for assessment of response duration and had a median duration of response of 12.0 months.
Nine of the 17 responders had a multi-lineage best response. Of the 14 patients who entered the extension, seven had improvement in more than one lineage following continuation of treatment: five patients with uni-lineage response improved to multi-lineage response (bi- or tri-lineage) and two patients with bi-lineage response improved to tri-lineage response. Three of the four bi-lineage responders also had meaningful improvements in hemoglobin (>1.5 g/dL); however, as their baseline hemoglobin was above 9 g/dL they are not counted as having an erythroid response.
The longest platelet transfusion free period in responders ranged from 8 to 1,190 days with a median of approximately 287 days. The longest RBC transfusion free period in responders ranged from 15 to 1,190 days with a median of approximately 266 days. Of the five patients who met protocol specified 'tri-lineage hematopoiesis' criteria for at least eight weeks and were tapered off Revolade, all five patients have maintained tri-lineage hematopoiesis since discontinuing treatment for a median follow up period of 20.6 months (range 5.7 to 22.5 months).
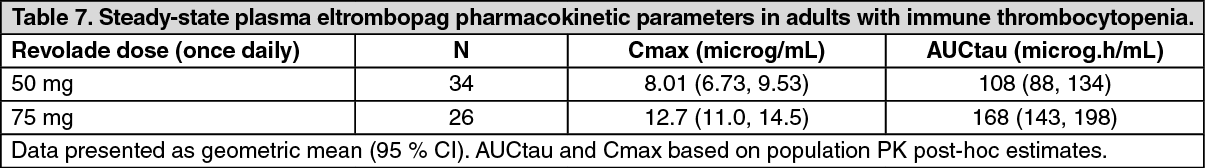
Pharmacokinetics (PK): The pharmacokinetic parameters of eltrombopag after administration of Revolade to adult patients with ITP are shown in Table 7. (See Table 7.)
Click on icon to see table/diagram/image
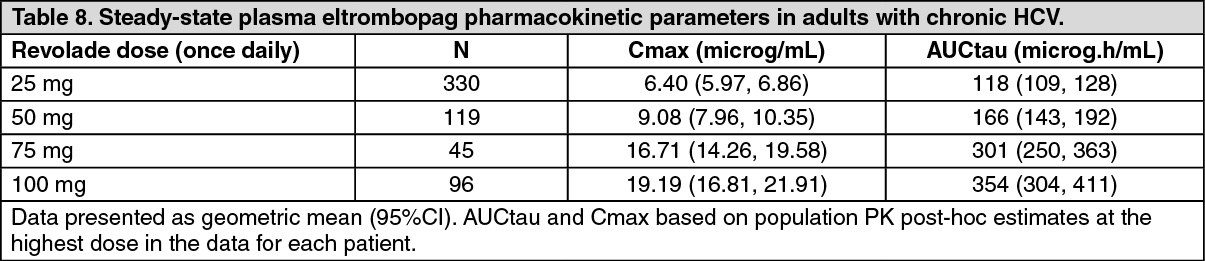
Plasma eltrombopag concentration-time data collected in 590 patients with HCV enrolled in Phase III studies TPL103922/ENABLE 1 and TPL108390/ENABLE 2 were combined with data from patients with HCV enrolled in the Phase II study TPL102357 and healthy adults in a population pharmacokinetic analysis. Plasma eltrombopag Cmax and AUCtau estimates for patients with HCV enrolled in the Phase III studies are presented for each dose studied in Table 8. A higher eltrombopag exposure was observed in patients with HCV at a given Revolade dose. (See Table 8.)
Click on icon to see table/diagram/image
The pharmacokinetic parameters of eltrombopag after administration of Revolade 150 mg to 45 patients with definitive immunosuppressive therapy-naïve severe aplastic anemia are shown in Table 9. (See Table 9.)
Click on icon to see table/diagram/image
Absorption: Eltrombopag is absorbed with a peak concentration occurring 2 to 6 hours after oral administration. Administration of Revolade concomitantly with antacids and other products containing polyvalent cations such as dairy products and mineral supplements significantly reduces eltrombopag exposure (see INTERACTIONS). In a relative bioavailability study in adults, Revolade powder for oral suspension delivered a 22% higher plasma AUCinf than the film-coated tablet formulation. The absolute oral bioavailability of eltrombopag after administration to humans has not been established. Based on urinary excretion and metabolites eliminated in feces, the oral absorption of drug-related material following administration of a single 75 mg Revolade solution dose was estimated to be at least 52%.
Distribution: Eltrombopag is highly bound to human plasma proteins (>99.9%). Eltrombopag is a substrate for BCRP, but is not a substrate for P-glycoprotein or OATP1B1.
Biotransformation/metabolism: Eltrombopag is primarily metabolized through cleavage, oxidation and conjugation with glucuronic acid, glutathione, or cysteine. In a human radiolabel study, eltrombopag accounted for approximately 64% of plasma radiocarbon AUCinf. Minor metabolites, each accounting for <10% of the plasma radioactivity, arising from glucuronidation and oxidation were also detected. Based on a human study with radiolabel eltrombopag, it is estimated that approximately 20% of a dose is metabolized by oxidation.
Elimination: Absorbed eltrombopag is extensively metabolized. The predominant route of eltrombopag excretion is via feces (59%) with 31% of the dose found in the urine as metabolites. Unchanged parent compound (eltrombopag) is not detected in urine. Unchanged eltrombopag excreted in feces accounts for approximately 20% of the dose. The plasma elimination half-life of eltrombopag is approximately 21-32 hours.
In vitro evaluation of drug interaction potential: Based on a human study with radiolabeled eltrombopag, glucuronidation plays a minor role in the metabolism of eltrombopag. Human liver microsome studies identified UGT1A1 and UGT1A3 as the enzymes responsible for eltrombopag glucuronidation. Eltrombopag was an inhibitor of a number of UGT enzymes
in vitro. Clinically significant drug interactions involving glucuronidation are not anticipated due to limited contribution of individual UGT enzymes in the glucuronidation of eltrombopag and potential co-medications.
Based on a human study with radiolabeled eltrombopag, approximately 21% of an eltrombopag dose could undergo oxidative metabolism. Human liver microsome studies identified CYP1A2 and CYP2C8 as the enzymes responsible for eltrombopag oxidation. In studies utilizing human liver microsomes, eltrombopag (up to 100 microM) showed no
in vitro inhibition of the CYP450 enzymes 1A2, 2A6, 2C19, 2D6, 2E1, 3A4/5, and 4A9/11 and was an inhibitor of CYP2C8 and CYP2C9 as measured using paclitaxel and diclofenac as the probe substrates, with IC50 values of 24.8 microM (11 microgram/mL) and 20.2 microM (8.9 microgram/mL), respectively.
In vitro studies demonstrate that eltrombopag is an inhibitor of the OATP1B1 transporter, with an IC
50 value of 2.7 microM (1.2 microg/mL) and an inhibitor of the BCRP transporter, with an IC
50 value of 2.7 microM (1.2 microg/mL).
In vitro studies identified CYP1A2 and CYP2C8 as the isoenzymes responsible for oxidative metabolism, uridine diphosphoglucuronyl transferase UGT1A1 and UGT1A3 as the isozymes responsible for glucuronidation, and that bacteria in the lower gastrointestinal tract may be responsible for the cleavage pathways.
In vitro studies demonstrated that eltrombopag is not a substrate for the organic anion transporter polypeptide, OATP1B1, but is an inhibitor of this transporter (IC
50 value of 2.7 microM (1.2 microgram/mL). In vitro studies also demonstrated that eltrombopag is a breast cancer resistance protein (BCRP) substrate and inhibitor (IC
50 value of 2.7 microM (1.2 microgram/mL)).
Special populations: Pediatric population (aged 1 to 17 years): The pharmacokinetics of eltrombopag have been evaluated in 168 pediatric ITP patients dosed once daily in two studies, TRA108062/PETIT and TRA115450/PETIT2. Plasma eltrombopag apparent clearance following oral administration (CL/F) increased with increasing body weight.
Approximately 30% lower plasma eltrombopag CL/F was observed in patients of East- /Southeast-Asian ancestry and 20% lower CL/F was observed in female patients. The bioavailability of the powder for oral suspension in pediatric patients was estimated as 29% lower than the film-coated tablet.
The pharmacokinetic parameters of eltrombopag in pediatric patients with ITP are shown in Table 10. (See Table 10.)
Click on icon to see table/diagram/image
Geriatric patients (60 years of age or above): The age difference of eltrombopag pharmacokinetics was evaluated using population pharmacokinetic analysis in 28 healthy subjects and 635 patients with HCV ranging from 19 to 74 years old. Based on model estimates, elderly (>60 years) patients had approximately 36% higher plasma eltrombopag AUCtau as compared to younger patients.
Gender: The influence of gender on the pharmacokinetics of eltrombopag was evaluated using a population pharmacokinetic analysis in 111 healthy adults (14 females) and 88 patients with ITP (57 females). Based on estimates from the population pharmacokinetic analysis, female ITP patients had approximately 50% higher plasma eltrombopag AUCtau as compared to male ITP patients, without adjustment for body weight differences.
The influence of gender on eltrombopag pharmacokinetics was evaluated using a population pharmacokinetic analysis in 663 patients with HCV (260 females). Based on model estimates, female HCV patients had approximately 41% higher plasma eltrombopag AUCtau as compared to male patients.
Race/Ethnicity: ITP: The influence of East-Asian ethnicity on the pharmacokinetics of eltrombopag was evaluated using a population PK analysis in 111 healthy adults (31 East-Asians) and 88 patients with ITP (18 East-Asians). Based on estimates from the population pharmacokinetic analysis, East-Asian ITP patients had approximately 87% higher plasma eltrombopag AUCtau values as compared to non-East-Asian patients who were predominantly Caucasian, without adjustment for body weight differences (see DOSAGE & ADMINISTRATION).
HCV-associated thrombocytopenia: The influence of East-/Southeast-Asian ethnicity on the pharmacokinetics of eltrombopag was evaluated using a population pharmacokinetic analysis in 663 patients with HCV (214 East-/Southeast-Asian). Based on estimates from the population pharmacokinetic analysis, East-/Southeast-Asian patients had similar pharmacokinetics of eltrombopag. On average, East-/Southeast-Asian patients had approximately 55% higher plasma eltrombopag AUCtau values as compared to patients of other races who were predominantly Caucasian (see DOSAGE & ADMINISTRATION).
Renal impairment: The pharmacokinetics of eltrombopag have been studied after administration of Revolade to adult patients with renal impairment. Following administration of a single 50 mg-dose, the AUCinf of eltrombopag was decreased by 32% (90% CI: 63% decrease, 26% increase) in patients with mild renal impairment, 36% (90% CI: 66% decrease, 19% increase) in patients with moderate renal impairment, and 60% (90% CI: 18% decrease, 80% decrease) in patients with severe renal impairment compared with healthy volunteers. There was a trend for reduced plasma eltrombopag exposure in patients with renal impairment, but there was substantial variability and significant overlap in exposures between patients with renal impairment and healthy volunteers.
Hepatic impairment: The pharmacokinetics of eltrombopag have been studied after administration of Revolade to adult patients with liver cirrhosis (hepatic impairment). Following the administration of a single 50-mg dose, the AUCinf of eltrombopag was increased by 41% (90% CI: 13% decrease, 128% increase) in patients with mild hepatic impairment, 93% (90% CI: 19%, 213%) in patients with moderate hepatic impairment, and 80% (90% CI: 11%, 192%) in patients with severe hepatic impairment compared with healthy volunteers. There was substantial variability and significant overlap in exposures between patients with hepatic impairment and healthy volunteers.
The influence of hepatic impairment on the pharmacokinetics of eltrombopag following repeat administration was evaluated using a population pharmacokinetic analysis in 28 healthy adults and 79 patients with chronic liver disease. Based on estimates from the population pharmacokinetic analysis, patients with liver cirrhosis (hepatic impairment) had higher plasma eltrombopag AUCtau values as compared to healthy volunteers, and AUCtau increased with increasing Child-Pugh score. Compared to healthy volunteers, patients with mild hepatic impairment had approximately 87% to 110% higher plasma eltrombopag AUCtau values and patients with moderate hepatic impairment had approximately 141% to 240% higher plasma eltrombopag AUCtau values.
A similar analysis was also conducted in 28 healthy adults and 635 patients with HCV. A majority of patients had Child Pugh score of 5-6. Based on estimates from the population pharmacokinetic analysis, patients with HCV had higher plasma eltrombopag AUCtau values as compared to healthy subjects, and AUCtau increased with increasing Child-Pugh score, HCV patients with mild hepatic impairment had approximately 100% to 144% higher plasma eltrombopag AUCtau compared with healthy subjects. For patients with HCV, Revolade should be initiated at a dose of 25 mg once daily (see Special populations (all indications): HEPATIC IMPAIRMENT under DOSAGE & ADMINISTRATION).
Toxicology: Non-Clinical Safety Data: Safety pharmacology and repeat dose toxicity: Treatment-related cataracts were detected in rodents and were dose and time-dependent. At ≥6 times the human clinical exposure based on AUC in ITP patients at 75 mg/day and 3 times the human clinical exposure based on AUC in HCV patients at 100 mg/day, cataracts were observed in mice after 6 weeks and rats after 28 weeks of dosing. At ≥4 times the human clinical exposure based on AUC in ITP patients at 75 mg/day and 2 times the human clinical exposure based on AUC in HCV patients at 100 mg/day, cataracts were observed in mice after 13 weeks and in rats after 39 weeks of dosing. At non-tolerated doses in pre-weaning juvenile rats dosed from Days 4-32 (approximately equating to a 2-year old human at the end of the dosing period), ocular opacities were observed (histology not performed) at 9 times the maximum human clinical exposure in pediatric ITP patients at 75 mg/day, based on AUC. However, cataracts were not observed in juvenile rats given tolerated doses at 5 times the human clinical exposure in pediatric ITP patients, based on AUC. Cataracts have not been observed in dogs after 52 weeks of dosing at 2 times the human clinical exposure in ITP patients at 75 mg/day and equivalent to the human clinical exposure in HCV patients at 100 mg/day, based on AUC (see PRECAUTIONS).
Renal tubular toxicity was observed in studies of up to 14 days duration in mice and rats at exposures that were generally associated with morbidity and mortality. Tubular toxicity was also observed in a 2 year oral carcinogenicity study in mice at doses of 25, 75 and 150 mg/kg/day. Effects were less severe at lower doses and were characterized by a spectrum of regenerative changes. The exposure at the lowest dose was 1.2 times the human clinical exposure based on AUC in ITP patients at 75 mg/day and 0.6 times the human clinical exposure based on AUC in HCV patients at 100 mg/day. Renal effects were not observed in rats after 28 weeks or in dogs after 52 weeks at exposures 4 and 2 times the human clinical exposure in ITP patients at 75 mg/day, respectively and 2 times and equivalent to the human clinical exposure in HCV patients at 100 mg/day, respectively based on AUC.
Carcinogenicity and mutagenicity: Eltrombopag was not carcinogenic in mice at doses up to 75 mg/kg/day or in rats at doses up to 40 mg/kg/day (exposures up to 4 and 5 times the human clinical exposure based on AUC in ITP patients at 75 mg/day and 2 times the human clinical exposure based on AUC in HCV patients at 100 mg/day). Eltrombopag was not mutagenic or clastogenic in a bacterial mutation assay or in two
in vivo assays in rats (micronucleus and unscheduled DNA synthesis, 10 times the human clinical exposure, based on Cmax in ITP patients at 75 mg/day and 7 times the human clinical exposure in HCV patients at 100 mg/day). In the
in vitro mouse lymphoma assay, eltrombopag was marginally positive (<3-fold increase in mutation frequency). These
in vitro and
in vivo findings suggest that eltrombopag does not pose a genotoxic risk to humans.
Reproductive toxicity: Eltrombopag did not affect female fertility in rats at doses up to 20 mg/kg/day (2 times the human clinical exposure based on AUC in patients with ITP at 75 mg/day and similar to the human clinical exposure based on AUC in patients with chronic hepatitis C at 100 mg/day). Eltrombopag did not affect male fertility in rats at doses up to 40 mg/kg/day, the highest dose tested (3 times the human clinical exposure based on AUC in patients with ITP at 75 mg/day and 2 times the human clinical exposure based on AUC in patients with chronic hepatitis C at 100 mg/day) (see also FEMALES AND MALES OF REPRODUCTIVE POTENTIAL under Use in Pregnancy & Lactation).
Juvenile animal studies: At non-tolerated doses in pre-weaning rats, ocular opacities were observed. However, at tolerated doses, no ocular opacities were observed (see Safety pharmacology and repeat dose toxicity as previously mentioned). There are no findings in juvenile rats to suggest a greater risk of toxicity with eltrombopag treatment in pediatric vs. adult patients.



 Sign Out
Sign Out