


 Sign Out
Sign Out



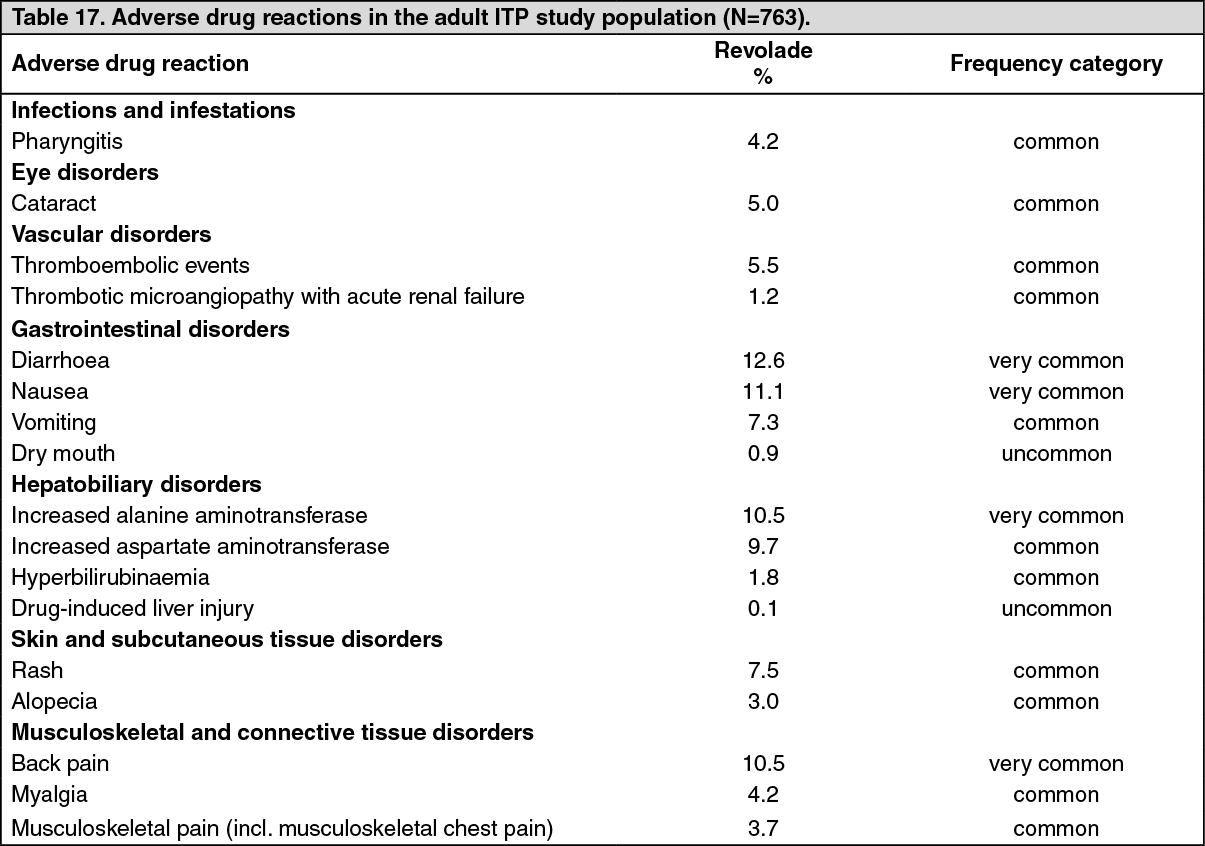
The most common adverse drug reactions (≥10%) for Revolade/Promacta were diarrhoea, nausea, increased alanine aminotransferase and back pain.
Immune thrombocytopenia in pediatric patients: The safety of Revolade was assessed in pediatric patients (aged 1 to 17 years) with previously treated ITP using the all-treated population from two studies (N=171) (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). PETIT2 (TRA115450) was a two-part, double-blind and open-label, randomized, placebo-controlled study. Patients were randomized 2:1 and received Revolade (n=63) or placebo (n=29) for up to 13 weeks in the randomized period of the study. PETIT (TRA108062) was a three-part, staggered cohort, open-label and double-blind, randomized, placebo-controlled study. Patients were randomized 2:1 and received Revolade (n=44) or placebo (n=21) for up to 7 weeks. Adverse drug reactions in the adult ITP study population (Table 17) may also occur in the pediatric ITP population. Additional adverse drug reactions occurring in the pediatric ITP study population (N=171) are shown in Table 18.
The most common additional adverse drug reactions (≥10%) for Revolade/Promacta were upper respiratory tract infection, pyrexia, abdominal pain, nasopharyngitis and cough.
Thrombocytopenia with HCV infection in adult patients: The safety of Revolade was assessed in adult patients treated with Revolade using two controlled studies, including data from patients who initially received Revolade in the pre-antiviral treatment phase and were later randomized to the placebo arm (N=1520) (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). ENABLE 1 (TPL103922; n=716, 715 treated with REVOLADE) and ENABLE 2 (TPL108390; n=805) were randomized, double-blind, placebo-controlled, multicenter studies to assess the efficacy and safety of Revolade in thrombocytopenic patients with HCV infection who were otherwise eligible to initiate antiviral therapy. In the HCV studies, the safety population consisted of all randomized patients who received double-blind study drug during Part 2 of ENABLE 1 (Revolade treatment n=449, placebo treatment n=232) and ENABLE 2 (Revolade treatment n = 506, placebo treatment n=252). Patients were analyzed according to the treatment received (total safety double-blind population, Revolade n=955 and placebo n=484). Adverse drug reactions for the HCV study population (N=1520) are shown in Table 19.
The most common adverse drug reactions (≥10%) for Revolade were anaemia, pyrexia, fatigue, headache, nausea, influenza like illness, diarrhoea, decreased appetite, asthenia, pruritus, cough, chills, and myalgia.
Definitive immunosuppressive therapy-naïve severe aplastic anemia in adult and pediatric patients: The safety of Revolade administered in combination with horse antithymocyte globulin (h-ATG) and cyclosporine to patients with severe aplastic anemia who had not received prior definitive immunosuppressive therapy (i.e., ATG therapy, alemtuzumab, or high dose cyclophosphamide) was evaluated in a single-arm, sequential cohort study (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). A total of 154 patients were enrolled and 153 were dosed in this study, of which 92 patients were enrolled to the cohort where Revolade, h-ATG, and cyclosporine were initiated concurrently at the recommended dose and schedule (Cohort 3 regimen): Revolade up to 150 mg once daily on Day 1 to Month 6 (D1M6) in combination with h-ATG on Days 1 to 4 and cyclosporine for 6 months, followed by low dose of cyclosporine (maintenance dose) for an additional 18 months for patients who achieved a hematologic response at 6 months. The median duration of exposure to Revolade in this cohort was 183 days with 83.7% of patients exposed for >12 weeks. Adverse drug reactions for the first-line SAA study population (N=92) are shown in Table 20.
The most common adverse drug reactions (≥10%) for Revolade were alanine aminotransferase increased, aspartate aminotransferase increased and blood bilirubin increased (including ocular icterus).
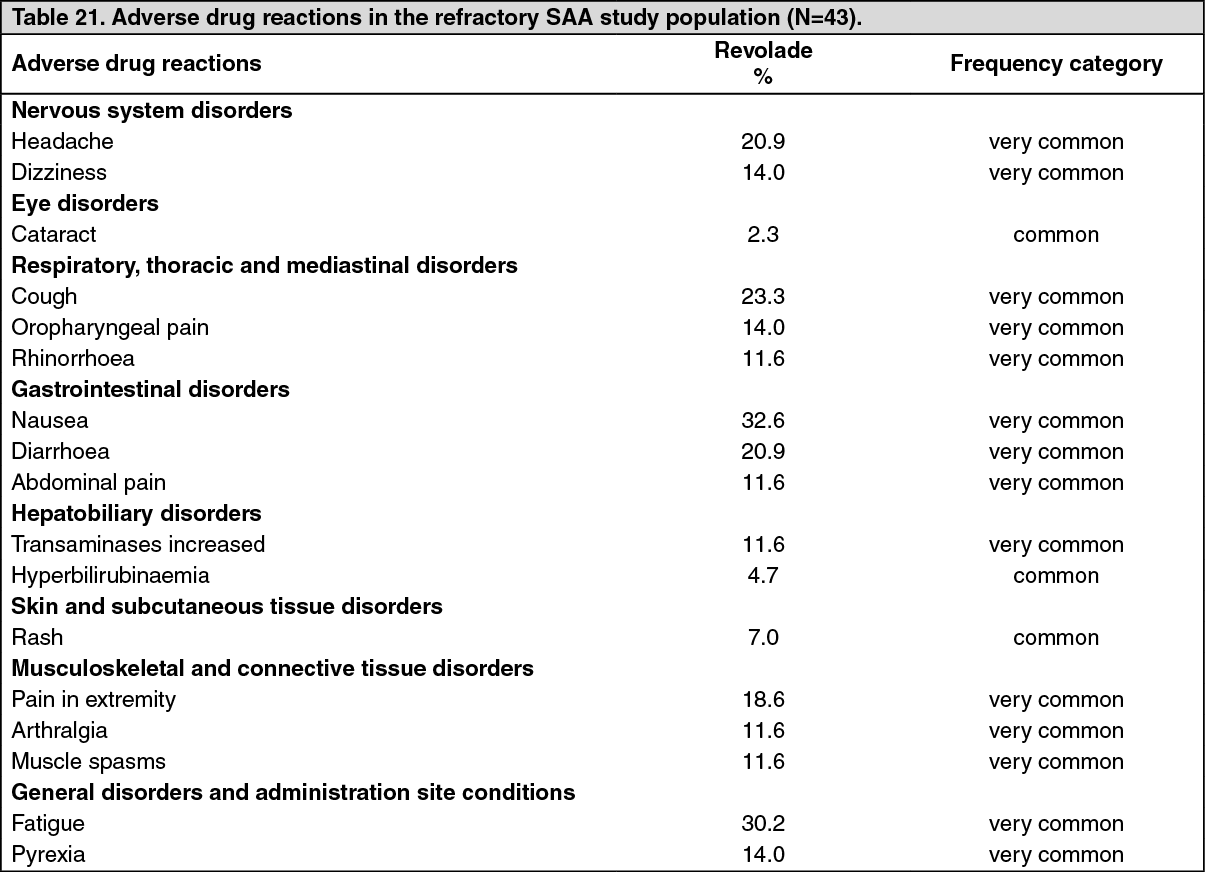
Refractory severe aplastic anemia in adult patients: The safety of Revolade in refractory severe aplastic anemia was assessed in a single-arm, open-label study (N=43) in which 11 patients (26%) were treated for >6 months and 7 patients (16%) were treated for >1 year (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). Adverse drug reactions for the refractory SAA study population (N=43) are shown in Table 21.
The most common adverse drug reactions (≥10%) for Revolade were nausea, fatigue, cough, headache, diarrhoea, pain in extremity, dizziness, oropharyngeal pain, pyrexia, rhinorrhoea, abdominal pain, transaminases increased, arthralgia and muscle spasms.
Most adverse drug reactions associated with Revolade in ITP, HCV and SAA were mild to moderate in severity, early in onset and rarely treatment-limiting.
Tabulated summary of reactions from clinical trials: Adverse drug reactions from clinical studies are listed as follows by MedDRA body system organ class and by frequency. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. The corresponding frequency category for each adverse drug reaction is based on the following convention (CIOMS III): very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000). (See Tables 17, 18, 19 and 20.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image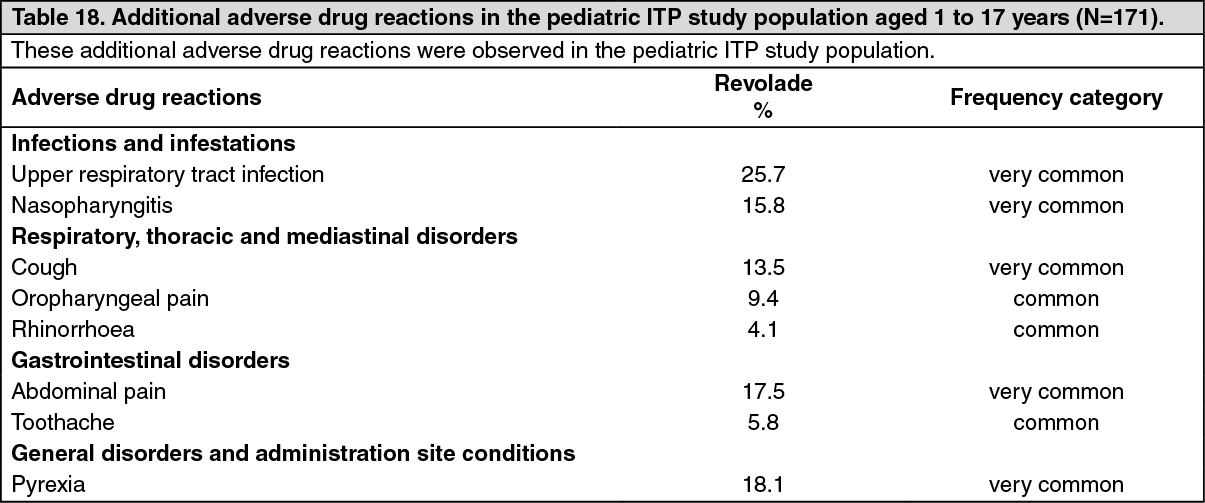 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image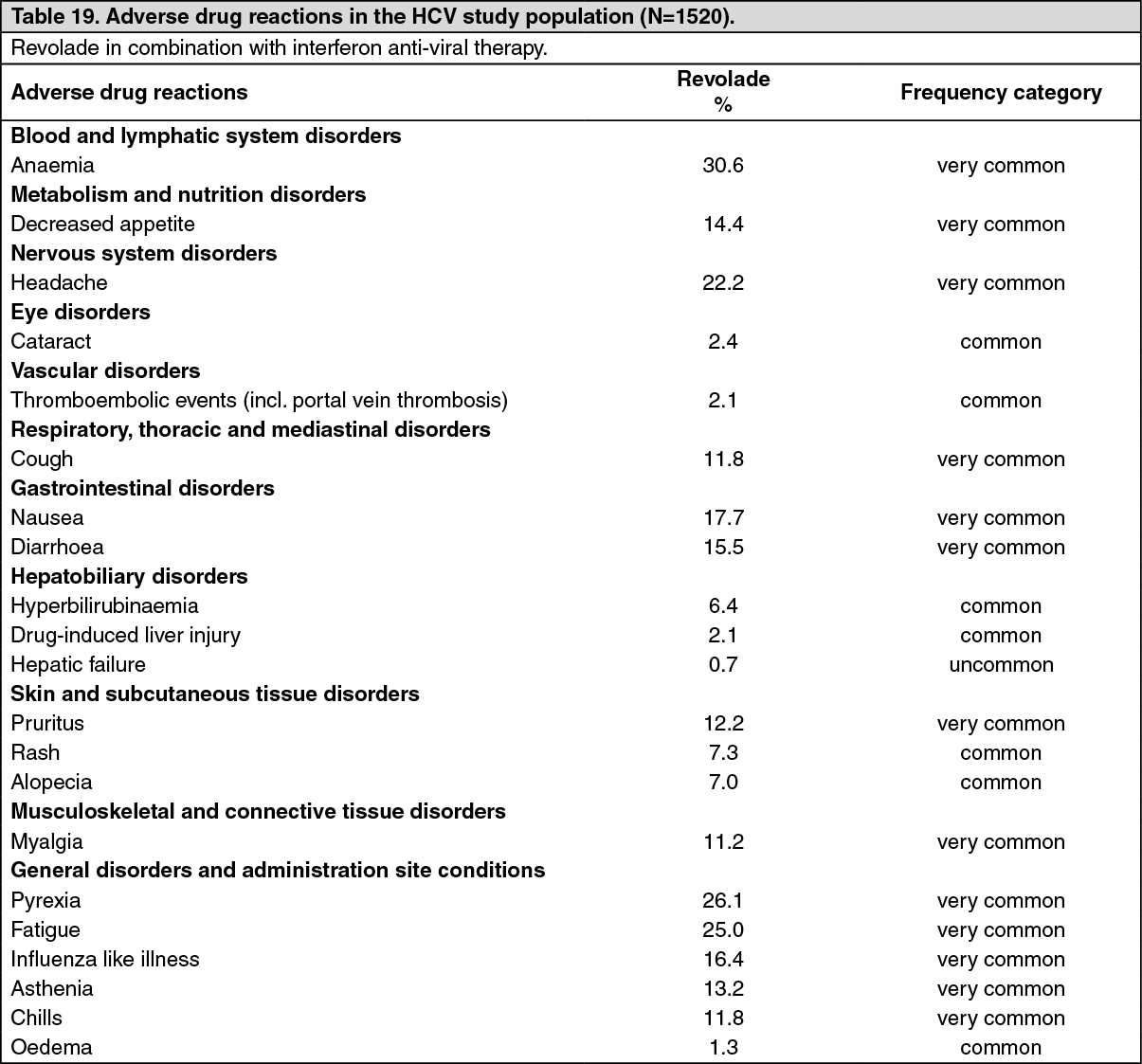 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image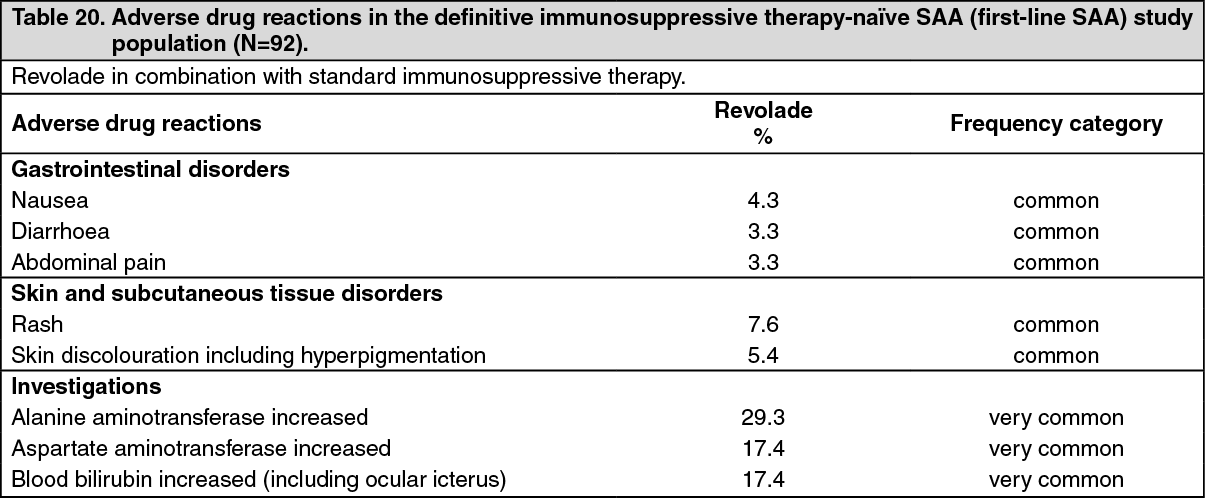 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageNew or worsening liver function laboratory abnormalities (CTCAE Grade 3 and Grade 4) in the Revolade D1-M6 cohort were 15.2% and 2.2% for AST, 26.4% and 4.3% for ALT, and 12.1% and 1.1% for bilirubin, respectively.
Pediatric patients: The safety assessment of Revolade in definitive immunosuppressive therapy-naïve pediatric SAA patients 2 to 17 years old is based on 37 patients enrolled in the single-arm, sequential cohort study: 2 patients aged 2 to 5 years, 12 patients aged 6 to 11 years, and 23 patients aged 12 to 17 years (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). The safety profile in pediatric patients was consistent with the safety profile observed in the overall population.
Cytogenetic abnormalities: In the single-arm study in patients with definitive immunosuppressive therapy-naïve SAA, patients had bone marrow aspirates evaluated for cytogenetic abnormalities. In the entire study across all cohorts, clonal cytogenetic evolution occurred in 15 out of 153 patients (10%). Of the 15 patients who experienced a cytogenetic abnormality, 7 patients had the loss of chromosome 7, six of which occurred within 6.1 months; 4 patients had chromosomal aberrations which were of unclear significance; 3 patients had a deletion of chromosome 13, which is considered a good prognostic factor in aplastic anemia; and 1 patient had a follow-up bone marrow assessment at 5 years with features of dysplasia with hypercellularity concerning for potential development of MDS. In the Revolade D1-M6 cohort, 7 patients had a new cytogenetic abnormality reported of which 4 had the loss of chromosome 7 occurring within 6.1 months.
It is unclear whether these findings occurred due to the underlying disease, the immunosuppressive therapy, and/or treatment with Revolade. (See Table 21.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the single-arm, open-label study in refractory SAA, patients had bone marrow aspirates evaluated for cytogenetic abnormalities. Eight patients had a new cytogenetic abnormality reported, including 5 patients who had changes in chromosome 7.
Adverse drug reactions from spontaneous reports and literature cases (frequency not known): The following adverse drug reactions have been reported during post-approval use of Revolade. These include spontaneous case reports as well as serious adverse events from registries, investigator sponsored studies, clinical pharmacology studies and exploratory studies in unapproved indications. Because they are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency, which is therefore categorized as not known. Adverse drug reactions are listed according to system organ classes in MedDRA. (See Table 22.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
View ADR Monitoring Form