


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Domperidone is a dopamine antagonist with anti-emetic properties. Domperidone does not readily cross the blood-brain barrier. In domperidone users, especially in adults, extrapyramidal side effects are very rare, but domperidone promotes the release of prolactin from the pituitary. Its anti-emetic effect may be due to a combination of peripheral (gastrokinetic) effects and antagonism of dopamine receptors in the chemoreceptor trigger zone, which lies outside the blood-brain barrier in the area postrema. Animal studies, together with the low concentrations found in the brain, indicate a predominantly peripheral effect of domperidone on dopamine receptors.
Studies in man have shown oral domperidone to increase lower esophageal pressure, improve antroduodenal motility and accelerate gastric emptying. There is no effect on gastric secretion.
Effect on QT/QTc Interval and Cardiac Electrophysiology: In accordance with ICH-E14 guidelines, a thorough QT study was performed in healthy subjects. This study included a placebo, active comparator and positive control and was conducted using recommended and supra-therapeutic doses (10 and 20 mg administered 4 times a day). This study found a maximal difference of QTc between domperidone and placebo in LS-means in the change from baseline of 3.4 msec for 20 mg domperidone administered 4 times a day on Day 4, and the 2-sided 90% CI (1.0 5.9 msec) did not exceed 10 msec. The QT prolongation observed in this study when domperidone was administered according to the recommended dosing regimen is not clinically relevant.
This lack of clinical relevance is corroborated by pharmacokinetics and QTc interval data from two older studies which involved a 5-day treatment of 20 mg and 40 mg domperidone administered 4 times a day. ECGs were recorded prior to the study, on Day 5 at 1 hour (approximately at tmax) after the morning dose, and 3 days later. In both studies, no difference between QTc after active treatment and placebo was observed. It was therefore concluded that domperidone administration of 80 and 160 mg daily doses had no clinically significant effect on QTc in healthy subjects.
Pharmacokinetics: Absorption: In fasting subjects, domperidone is rapidly absorbed after oral administration, with peak plasma concentrations occurring at approximately 60 minutes after dosing. The key pharmacokinetic parameters after a single or multiple doses (administered 4 times a day) of 10 mg domperidone base tablets to healthy subjects are presented in the table as follows. The Cmax and AUC values of domperidone increased proportionally with dose in the 10 mg to 20 mg dose range. (See Table 1.)
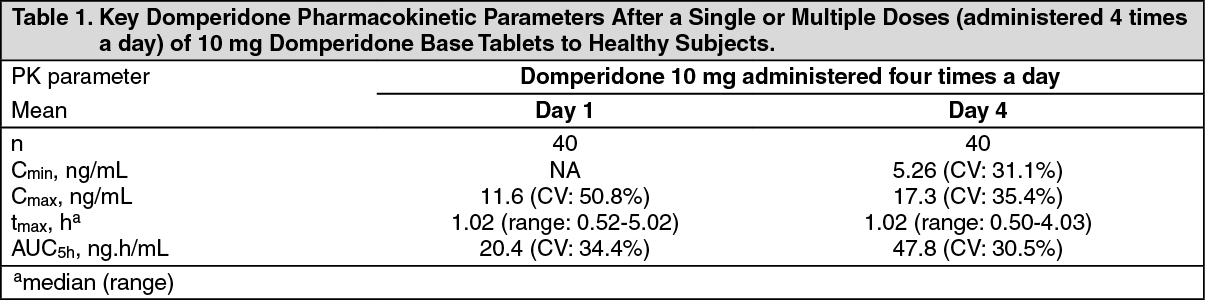 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe low absolute bioavailability of oral domperidone (approximately 15%) is due to an extensive first-pass metabolism in the gut wall and liver. Although domperidone's bioavailability is enhanced in normal subjects when taken after a meal, patients with gastro-intestinal complaints should take domperidone 15-30 minutes before a meal. Reduced gastric acidity impairs the absorption of domperidone base. Oral bioavailability of domperidone base is decreased by prior concomitant administration of cimetidine and sodium bicarbonate. The time of peak absorption is slightly delayed and the AUC somewhat increased when the oral drug is taken after a meal.
There are no pharmacokinetic data available for 30 mg suppositories. The bioavailability of a 60 mg suppository after single or repeated dosing is approximately 65% of 80 mg of oral tablets given over 24 hours. After rectal administration of 60 mg suppositories, mean domperidone plasma concentrations between 20 and 40 ng/mL are maintained from approximately 0.5 to 5 hours after single- and multiple-dose administration. Following single-dose administration, mean peak plasma levels of 60 mg suppositories are 89% of that of two 10 mg oral tablets, but the mean dose-normalized rectal bioavailability relative to oral tablets is 64%. Following multiple-dose administration, mean peak plasma levels and dose normalized bioavailability of 60 mg suppositories administered every 12 hours are 63% and 66%, respectively, of two 10 mg oral tablets administered every 6 hours.
Distribution: Domperidone is 91-93% bound to plasma proteins. Distribution studies with radiolabelled drug in animals have shown wide tissue distribution, but low brain concentration. Small amounts of drug cross the placenta in rats.
Metabolism: Domperidone undergoes rapid and extensive hepatic metabolism by hydroxylation and N-dealkylation. In vitro metabolism experiments with diagnostic inhibitors revealed that CYP3A4 is a major form of cytochrome P-450 involved in the N-dealkylation of domperidone, whereas CYP3A4, CYP1A2 and CYP2E1 are involved in domperidone aromatic hydroxylation.
Excretion: Urinary and fecal excretions amount to 31 and 66% of the oral dose, respectively. The proportion of the drug excreted unchanged is small (10% of fecal excretion and approximately 1% of urinary excretion).
The plasma half-life after a single oral dose is 7-9 hours in healthy subjects, but is prolonged in patients with severe renal insufficiency.
Special Populations: Hepatic impairment: In subjects with moderate hepatic impairment (Pugh score 7 to 9, Child-Pugh rating B), the AUC and Cmax of domperidone is 2.9- and 1.5-fold higher, respectively, than in healthy subjects. The unbound fraction is increased by 25%, and the terminal elimination half-life is prolonged from 15 to 23 hours. Subjects with mild hepatic impairment have a somewhat lower systemic exposure than healthy subjects based on Cmax and AUC, with no change in protein binding or terminal half-life. Subjects with severe hepatic impairment were not studied (see Contraindications).
Renal impairment: In subjects with severe renal insufficiency (serum creatinine > 6 mg/100 mL, i.e. > 0.6 mmol/L) the half-life of domperidone is increased from 7.4 to 20.8 hours, but plasma drug levels are lower than in subjects with normal renal function. Very little unchanged drug (approximately 1%) is excreted via the kidneys (see Dosage & Administration).
Pediatric patients: Based on limited pharmacokinetic data, domperidone plasma concentrations in preterm neonates were consistent with those reported in adults.
Toxicology: Non-Clinical Information: At a high, maternally toxic dose of 200 mg/kg/day, teratogenic effects (organ abnormalities such as anophthalmia, microphthalmia and displacement of the subclavian artery) were seen in the rat. The clinical significance of these findings is unknown. No teratogenicity was observed in mice and rabbits.
Electrophysiological in vitro and in vivo studies have shown that domperidone, at high concentrations, may prolong the QTc interval.
In juvenile rats, a no observed adverse effect level of 10 mg/kg was observed following 30 days of once daily repeat intraperitoneal dosing. Single intraperitoneal or intravenous doses showed similar LD50 values (mean range 53-76 mg/kg) in both juvenile and adult rats.