Posology: Treatment of symptomatic anaemia in adult and paediatric chronic renal failure patients: In patients with chronic renal failure where intravenous access is routinely available (haemodialysis patients) administration by the intravenous route is preferable. Where intravenous access is not readily available (patients not yet undergoing dialysis and peritoneal dialysis patients) Alvoetin may be administered subcutaneously.
Anaemia symptoms and sequelae may vary with age, gender, and co-morbid medical condition physician's evaluation of the individual patient's clinical course and condition is necessary.
Alvoetin should be administered in order to increase haemoglobin to not greater than 12 g/dl (7.5 mmol/l). A rise in haemoglobin of greater than 2 g/dl (1.25 mmol/l) over a four week period should be avoided. If it occurs, appropriate dose adjustment should be made as provided.
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed through dose management, with consideration for the haemoglobin target range of 10 g/dl (6.2 mmol/l) to 12 g/dl (7.5 mmol/l). In paediatric patients the recommended target haemoglobin range is between 9.5 and 11 g/dl (5.9-6.8 mmol/l).
A sustained haemoglobin level of greater than 12 g/dl (7.5 mmol/l) should be avoided. If the haemoglobin is rising by more than 2 g/dl (1.25 mmol/l) per month, or if the sustained haemoglobin exceeds 12 g/dl (7.5 mmol/l) reduce the epoetin alfa dose by 25%. If the haemoglobin exceeds 13 g/dl (8.1 mmol/l), discontinue therapy until it falls below 12 g/dl (7.5 mmol/l) and then reinstitute epoetin alfa therapy at a dose 25% below the previous dose.
Patients should be monitored closely to ensure that the lowest approved dose of Alvoetin is used to provide adequate control of anaemia and of the symptoms of anaemia.
Iron status should be evaluated prior to and during treatment and iron supplementation administered if necessary. In addition, other causes of anaemia, such as B
12 or folate deficiency, should be excluded before instituting therapy with epoetin alfa.
Non response to epoetin alfa therapy should prompt a search for causative factors. These include: iron, folate, or Vitamin B
12 deficiency; aluminium intoxication; intercurrent infections; inflammatory or traumatic episodes; occult blood loss; haemolysis; and bone marrow fibrosis of any origin.
Adult haemodialysis patients: In patients on haemodialysis where intravenous access is readily available, administration by the intravenous route is preferable.
The treatment is divided into two stages. Correction phase: 50 IU/kg, 3 times per week.
When a dose adjustment is necessary, this should be done in steps of at least four weeks. At each step, the increase or reduction in dose should be of 25 IU/kg, 3 times per week.
Maintenance phase: Dosage adjustment in order to maintain haemoglobin values at the desired level: Hb between 10 and 12 g/dl (6.2 - 7.5 mmol/l).
The recommended total weekly dose is between 75 and 300 IU/kg.
The clinical data available suggest that those patients whose initial haemoglobin is very low (< 6 g/dl or < 3.75 mmol/l) may require higher maintenance doses than those whose initial anaemia is less severe (> 8 g/dl or > 5 mmol/l).
Paediatric haemodialysis patients: The treatment is divided into two stages.
Correction phase: 50 IU/kg, 3 times per week by the intravenous route.
When a dose adjustment is necessary, this should be done in steps of 25 IU/kg, 3 times per week at intervals of at least 4 weeks until the desired goal is achieved.
Maintenance phase: Dosage adjustment in order to maintain haemoglobin values at the desired level: Hb between 9.5 and 11 g/dl (5.8 - 6.8 mmol/l).
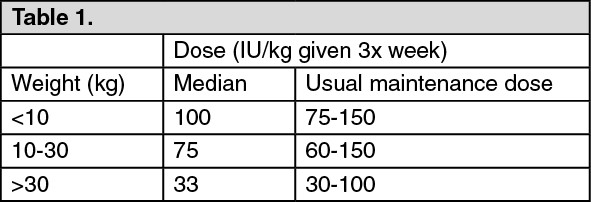
Generally, children under 30 kg require higher maintenance doses than children over 30 kg and adults. For example, the following maintenance doses were observed in clinical trials after 6 months of treatment. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The clinical data available suggest that those patients whose initial haemoglobin is very low (< 6.8 g/dl or < 4.25 mmol/l) may require higher maintenance doses than those whose initial haemoglobin is higher (> 6.8 g/dl or > 4.25 mmol/l).
Adult patients with renal insufficiency not yet undergoing dialysis: Where intravenous access is not readily available Alvoetin may be administered subcutaneously.
The treatment is divided into two stages.
Correction phase: Starting dose of 50 IU/kg, 3 times per week, followed if necessary by a dosage increase with 25 IU/kg increments (3 times per week) until the desired goal is achieved (this should be done in steps of at least four weeks).
Maintenance phase: Dosage adjustment in order to maintain haemoglobin values at the desired level: Hb between 10 and 12 g/dl (6.2 - 7.5 mmol/l) (maintenance dose between 17 and 33 IU/kg, 3 times per week).
The maximum dosage should not exceed 200 IU/kg, 3 times per week.
Adult peritoneal dialysis patients: Where intravenous access is not readily available Alvoetin may be administered subcutaneously.
The treatment is divided into two stages.
Correction phase: Starting dose of 50 IU/kg, 2 times per week.
Maintenance phase: Dosage adjustment in order to maintain haemoglobin values at the desired level: Hb between 10 and 12 g/dl (6.2 - 7.5 mmol/l) (maintenance dose between 25 and 50 IU/kg 2 times per week into 2 equal injections).
Treatment of patients with chemotherapy induced anaemia: Alvoetin should be administered by the subcutaneous route to patients with anaemia (e.g. haemoglobin concentration ≤ 10 g/dl (6.2 mmol/l)).
Anaemia symptoms and sequelae may vary with age, gender, and overall burden of disease; a physician's evaluation of the individual patient's clinical course and condition is necessary.
Due to intra-patient variability, occasional individual haemoglobin values for a patient above and below the desired haemoglobin level may be observed. Haemoglobin variability should be addressed through dose management, with consideration for the haemoglobin target range of 10 g/dl (6.2 mmol/l) to 12 g/dl (7.5 mmol/l). A sustained haemoglobin level of greater than 12 g/dl (7.5 mmol/l) should be avoided; guidance for appropriate dose adjustment for when haemoglobin values exceed 12 g/dl (7.5 mmol/l) is described as follows.
Epoetin alfa therapy should continue until one month after the end of chemotherapy.
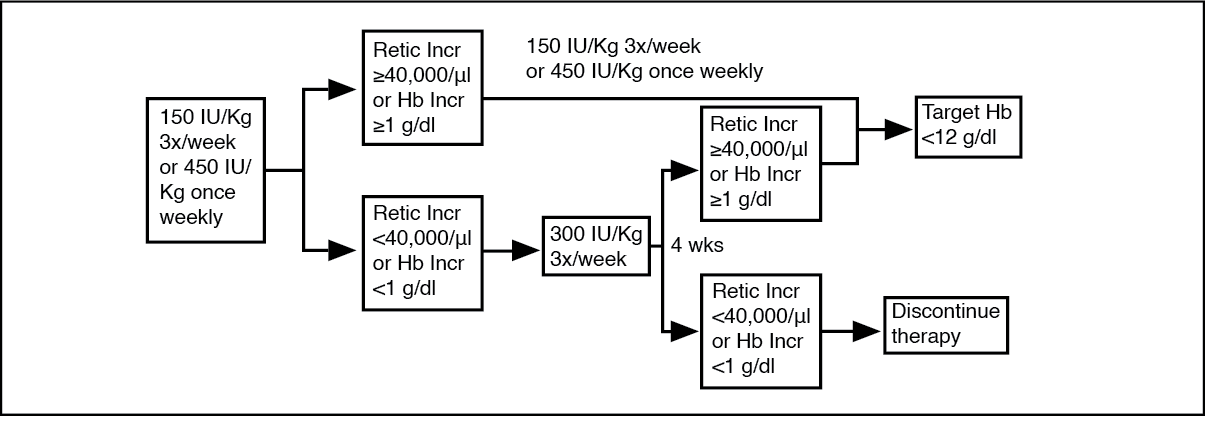
The initial dose is 150 IU/kg given subcutaneously 3 times per week. Alternatively, Alvoetin can be administered at an initial dose of 450 IU/kg subcutaneously once weekly.
If the haemoglobin has increased by at least 1 g/dl (0.62 mmol/l) or the reticulocyte count has increased ≥ 40,000 cells/µl above baseline after 4 weeks of treatment, the dose should remain at 150 IU/kg 3 times per week or 450 IU/kg once weekly.
If the haemoglobin increase is < 1 g/dl (< 0.62 mmol/l) and the reticulocyte count has increased < 40,000 cells/μl above baseline, increase the dose to 300 IU/kg 3 times per week.
If after an additional 4 weeks of therapy at 300 IU/kg 3 times per week, the haemoglobin has increased ≥ 1 g/dl (≥ 0.62 mmol/l) or the reticulocyte count has increased ≥ 40,000 cells/μl, the dose should remain at 300 IU/kg 3 times per week. However, if the haemoglobin has increased < 1 g/dl (< 0.62 mmol/l) and the reticulocyte count has increased < 40,000 cells/μl above baseline, response is unlikely and treatment should be discontinued.
The recommended dosing regimen is described in the following diagram. (See figure.)
Click on icon to see table/diagram/image
Patients should be monitored closely to ensure that the lowest approved dose of erythropoiesis-stimulating agent (ESA) is used to provide adequate control of the symptoms of anaemia.
Dose adjustment to maintain haemoglobin concentrations between 10 g/dl - 12 g/dl: If the haemoglobin is rising by more than 2 g/dl (1.25 mmol/l) per month, or if the haemoglobin exceeds 12 g/dl (7.5 mmol/l), reduce the epoetin alfa dose by about 25 - 50%.
If the haemoglobin exceeds 13 g/dl (8.1 mmol/l), discontinue therapy until it falls below 12 g/dl (7.5 mmol/l) and then reinstitute epoetin alfa therapy at a dose 25% below the previous dose.
Adult surgery patients in an autologous predonation programme: The intravenous route of administration should be used. At the time of donating blood, epoetin alfa should be administered after the completion of the blood donation procedure.
Mildly anaemic patients (haematocrit of 33-39%) requiring predeposit of ≥ 4 units of blood should be treated with epoetin alfa at 600 IU/kg, 2 times weekly for 3 weeks prior to surgery. Using this regimen, it was possible to withdraw ≥ 4 units of blood from 81% of epoetin alfa-treated patients compared to 37% of placebo-treated patients. Epoetin alfa therapy reduced the risk of exposure to homologous blood by 50% compared to patients not receiving epoetin alfa.
All patients being treated with epoetin alfa should receive adequate iron supplementation (e.g. 200 mg oral elemental iron daily) throughout the course of epoetin alfa treatment. Iron supplementation should be started as soon as possible, even several weeks prior to initiating the autologous predeposit, in order to achieve high iron stores prior to starting epoetin alfa therapy.
Adult patients scheduled for major elective orthopaedic surgery: The subcutaneous route of administration should be used.
The recommended dose regimen is 600 IU/kg of epoetin alfa, given weekly for three weeks (days -21, -14 and -7) prior to surgery and on the day of surgery. In cases where there is a medical need to shorten the lead time before surgery to less than three weeks, 300 IU/kg epoetin alfa should be given daily for 10 consecutive days prior to surgery, on the day of surgery and for four days immediately thereafter. When performing haematologic assessments during the preoperative period, if the haemoglobin level reaches 15 g/dl, or higher, administration of epoetin alfa should be stopped and further dosages should not be given.
Care should be taken to ensure that at the outset of the treatment patients are not iron deficient. All patients being treated with epoetin alfa should receive adequate iron supplementation (e.g. 200 mg oral elemental iron daily) throughout the course of epoetin alfa treatment. If possible, iron supplementation should be started prior to epoetin alfa therapy, to achieve adequate iron stores.
Zidovudine-treated HIV-infected Patients: Prior to beginning epoetin alfa, it is recommended that the endogenous serum erythropoietin level be determined (prior to transfusion). Available evidence suggests that patients receiving zidovudine with endogenous serum erythropoietin levels > 500 mIU/mL are unlikely to respond to therapy with epoetin alfa.
In zidovudine-treated HIV-infected patients the dosage of epoetin alfa should be titrated for each patient to achieve and maintain the lowest hemoglobin level sufficient to avoid the need for blood transfusion and not to exceed the upper safety limit of 12 g/dL.
Starting Dose: For adult patients with serum erythropoietin levels <= 500 mIU/ml who are receiving a dose of zidovudine <= 4200 mg/week, the recommended starting dose of epoetin alfa is 100 IU/kg as an i.v. or s.c. injection three times per week for 8 weeks.
Increase Dose: During the dose adjustment phase of therapy, the hemoglobin should be monitored weekly. If the response is not satisfactory in terms of reducing transfusion requirements or increasing hemoglobin after 8 weeks of therapy, the dose of epoetin alfa can be increased by 50 to 100 IU/kg three times a week. Response should be evaluated every 4 to 8 weeks thereafter and the dose adjusted accordingly by 50 to 100 IU/kg increments three times a week. If patients have not responded satisfactorily to an epoetin alfa dose of 300 IU/kg three times a week, it is unlikely they will respond to higher doses of epoetin alfa.
Maintenance Dose: After attainment of the desired response (i.e. reduced transfusion requirements or increased hemoglobin), the dose of epoetin alfa should be titrated to maintain the response based on factors such as variations in zidovudine dose and the presence of intercurrent infectious or inflammatory episodes. If the hemoglobin exceeds the upper safety limit of 12 g/dL, the dose should be discontinued until the hemoglobin drops below 11 g/dL. The dose should be reduced by 25% when treatment is resumed and then titrated to maintain the desired hemoglobin.
Method of administration: As with any other injectable product, check that there are no particles in the solution or change in colour.
Intravenous injection: Over at least one to five minutes, depending on the total dose.
In haemodialysed patients, a bolus injection may be given during the dialysis session through a suitable venous port in the dialysis line. Alternatively, the injection can be given at the end of the dialysis session via the fistula needle tubing, followed by 10 ml of isotonic saline to rinse the tubing and ensure satisfactory injection of the product into the circulation.
A slower injection is preferable in patients who react to the treatment with "flu-like" symptoms. Do not administer by intravenous infusion or mixed with other drugs.
Subcutaneous injection: A maximum volume of 1 ml at one injection site should generally not be exceeded. In case of larger volumes, more than one site should be chosen for the injection.
The injections are given in the limbs or the anterior abdominal wall.
In those situations in which the physician determines that a patient or caregiver can safely and effectively administer Alvoetin subcutaneously, instruction as to the proper dosage and administration should be provided.



 Sign Out
Sign Out