Pharmacotherapeutic group: Antineoplastic agent, protein kinase inhibitors.
ATC code: L01ED04.
Pharmacology: Pharmacodynamics: Mechanism of Action: Brigatinib is a tyrosine kinase inhibitor that targets ALK, c-ros oncogene 1 (ROS1), and insulin-like growth factor 1 receptor (IGF-1R). Brigatinib inhibited autophosphorylation of ALK and ALK-mediated phosphorylation of the downstream signalling protein STAT3 in in vitro and in vivo assays.
Brigatinib inhibited the in vitro proliferation of cell lines expressing EML4-ALK and NPM-ALK fusion proteins and demonstrated dose-dependent inhibition of EML4-ALK-positive NSCLC xenograft growth in mice. Brigatinib inhibited the in vitro and in vivo viability of cells expressing mutant forms of EML4-ALK associated with resistance to ALK inhibitors, including G1202R and L1196M.
Cardiac Electrophysiology: In Study 101, the QT interval prolongation potential of Alunbrig was assessed in 123 patients with advanced malignancies following once daily brigatinib doses of 30 mg to 240 mg. The maximum mean QTcF (corrected QT by the Fridericia method) change from baseline was less than 10 msec. An exposure-QT analysis suggested no concentration-dependent QTc interval prolongation.
Clinical efficacy and safety: ALTA 1L: The safety and efficacy of Alunbrig was evaluated in a randomised (1:1), open-label, multicentre trial (ALTA 1L) in 275 adult patients with advanced ALK-positive NSCLC who had not previously received an ALK-targeted therapy. Eligibility criteria permitted enrolment of patients with a documented ALK rearrangement based on a local standard of care testing and an ECOG Performance status of 0-2. Patients were allowed to have up to 1 prior regimen of chemotherapy in the locally advanced or metastatic setting. Neurologically stable patients with treated or untreated central nervous system (CNS) metastases, including leptomeningeal metastases, were eligible. Patients with a history of pulmonary interstitial disease, drug-related pneumonitis, or radiation pneumonitis were excluded.
Patients were randomised in a 1:1 ratio to receive Alunbrig 180 mg once daily with a 7-day lead-in at 90 mg once daily (N = 137) or crizotinib 250 mg orally twice daily (N = 138). Randomisation was stratified by brain metastases (present, absent) and prior chemotherapy use for locally advanced or metastatic disease (yes, no).
Patients in the crizotinib arm who experienced disease progression were offered crossover to receive treatment with Alunbrig. Among all 121 patients who were randomised to the crizotinib arm and discontinued study treatment by the time of the final analysis, 99 (82%) patients received subsequent ALK tyrosine kinase inhibitors (TKIs). Eighty (66%) patients who were randomised to the crizotinib arm received subsequent Alunbrig treatment, including 65 (54%) patients who crossed over in the study.
The major outcome measure was progression-free survival (PFS) according to Response Evaluation Criteria in Solid Tumours (RECIST v1.1) as evaluated by a Blinded Independent Review Committee (BIRC). Additional outcome measures as evaluated by the BIRC include confirmed objective response rate (ORR), duration of response (DOR), time to response, disease control rate (DCR), intracranial ORR, intracranial PFS, and intracranial DOR. Investigator-assessed outcomes include PFS and overall survival.
Baseline demographics and disease characteristics in ALTA 1L were median age 59 years old (range 27 to 89; 32% 65 and over), 59% White and 39% Asian, 55% female, 39% ECOG PS 0, and 56% ECOG PS 1, 58% never smokers, 93% Stage IV disease, 96% adenocarcinoma histology, 30% CNS metastases at baseline, 14% prior radiotherapy to the brain, and 27% prior chemotherapy. Sites of extra-thoracic metastases include brain (30% of patients), bone (31% of patients), and liver (20% of patients). The median relative dose intensity was 97% for Alunbrig and 99% for crizotinib.
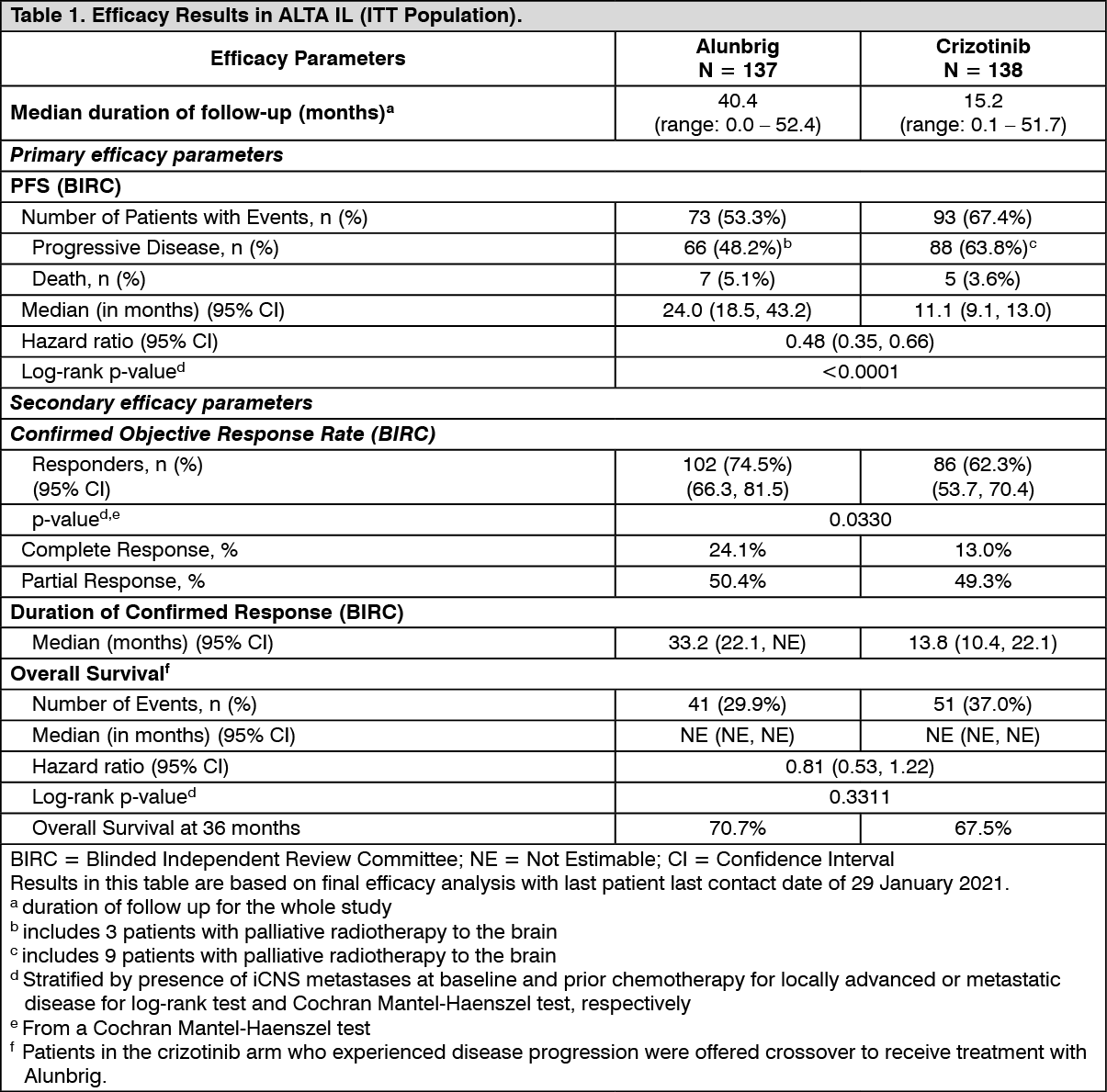
At the primary analysis performed at a median follow-up duration of 11 months in the Alunbrig arm, the ALTA 1L study met its primary endpoint demonstrating a statistically significant improvement in PFS by BIRC.
A protocol-specified efficacy analysis with cut-off date of 28 June 2019 was performed at a median follow-up duration of 24.9 months in the Alunbrig arm. The median PFS by BIRC in the ITT population was 24 months in the Alunbrig arm and 11 months in the crizotinib arm (HR =0.49 [95% CI (0.35, 0.68)], p <0.0001).
The results from the protocol-specified final analysis with last patient last contact date of 29 January 2021 performed at a median follow-up duration of 40.4 months in the Alunbrig arm are presented as follows. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
BIRC assessment of intracranial efficacy according to RECIST v1.1 in patients with any brain metastases and patients with measurable brain metastases (≥10 mm in longest diameter) at baseline are summarized in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
ALTA: The safety and efficacy of Alunbrig was evaluated in a randomised (1:1), open-label, multicenter trial (ALTA) in 222 adult patients with locally advanced or metastatic ALK-positive NSCLC who had progressed on crizotinib. Eligibility criteria permitted enrolment of patients with a documented ALK rearrangement based on a validated test, ECOG Performance Status of 0-2, and prior chemotherapy. Additionally, patients with central nervous system (CNS) metastases were included, provided they were neurologically stable and did not require an increasing dose of corticosteroids. Patients with a history of pulmonary interstitial disease or drug-related pneumonitis were excluded.
Patients were randomised in a 1:1 ratio to receive Alunbrig either 90 mg once daily (90 mg regimen, N = 112) or 180 mg once daily with 7-day lead-in at 90 mg once daily (180 mg regimen, N = 110). The median duration of follow-up was 22.9 months. Randomisation was stratified by brain metastases (present, absent) and best prior response to crizotinib therapy (complete or partial response, any other response/unknown).
The major outcome measure was confirmed objective response rate (ORR) according to Response Evaluation Criteria in Solid Tumours (RECIST v1.1) as evaluated by investigator. Additional outcome measures included confirmed ORR as evaluated by an Independent Review Committee (IRC); time to response; progression free survival (PFS); duration of response (DOR); overall survival; and intracranial ORR and intracranial DOR as evaluated by an IRC.
Baseline demographics and disease characteristics in ALTA were median age 54 years old (range 18 to 82; 23% 65 and over), 67% White and 31% Asian, 57% female, 36% ECOG PS 0 and 57% ECOG PS 1, 7% ECOG PS2, 60% never smoker, 35% former smoker, 5% current smoker, 98% Stage IV, 97% adenocarcinoma, and 74% prior chemotherapy. The most common sites of extra-thoracic metastasis included 69% brain (of whom 62% had received prior radiation to the brain), 39% bone, and 26% liver.
Efficacy results from ALTA analysis are summarised in Table 3 and the Kaplan-Meier (KM) curve for investigator-assessed PFS is shown in Figure 2. (See Table 3 and Figure 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
IRC assessments of intracranial ORR and duration of intracranial response in patients from ALTA with measurable brain metastases (≥ 10 mm in longest diameter) at baseline are summarised in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
In patients with any brain metastases at baseline, intracranial disease control rate was 77.8% (95% CI 67.2-86.3) in the 90 mg arm (N = 81) and 85.1% (95% CI 75-92.3) in the 180 mg arm (N = 74).
Study 101: In a separate dose finding study, 25 patients with ALK-positive NSCLC that progressed on crizotinib were administered Alunbrig at 180 mg once daily with 7-day lead-in at 90 mg once daily regimen. Of these, 19 patients had an investigator-assessed confirmed objective response (76%; 95% CI: 55, 91) and the KM estimate median duration of response among the 19 responders was 26.1 months (95% CI: 7.9, 26.1). The KM median PFS was 16.3 months (95% CI: 9.2, NE) and the 12-month probability of overall survival was 84.0% (95% CI: 62.8, 93.7).
Paediatric population: The European Medicines Agency has waived the obligation to submit the results of studies with Alunbrig in all subsets of the paediatric population in lung carcinoma (small cell and non-small cell carcinoma) (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Absorption: In Study 101, following administration of a single oral dose of brigatinib (30-240 mg) in patients, the median time to peak concentration (T
max) was 1-4 hours postdose. After a single dose and at steady state, systemic exposure was dose proportional over the dose range of 60-240 mg once daily. Modest accumulation was observed upon repeated dosing (geometric mean accumulation ratio: 1.9 to 2.4). The geometric mean steady state C
max of brigatinib at doses of 90 mg and 180 mg once daily was 552 and 1,452 ng/mL, respectively, and the corresponding AUC
0-τ was 8,165 and 20,276 h∙ng/mL, respectively. Brigatinib is a substrate of the transporter proteins P-gp and BCRP.
In healthy subjects, compared to overnight fasting, a high fat meal reduced brigatinib C
max by 13% with no effect on AUC. Brigatinib can be administered with or without food.
Distribution: Brigatinib was moderately bound (91%) to human plasma proteins and binding was not concentration-dependent. The blood-to-plasma concentration ratio is 0.69. In patients given brigatinib 180 mg once daily, the geometric mean apparent volume of distribution (V
z/F) of brigatinib at steady state was 307 L, indicating moderate distribution into tissues.
Biotransformation: In vitro studies demonstrated that brigatinib is primarily metabolised by CYP2C8 and CYP3A4, and to a much lesser extent by CYP3A5.
Following oral administration of a single 180 mg dose of [
14C]brigatinib to healthy subjects, N-demethylation and cysteine conjugation were the two major metabolic clearance pathways. In urine and faeces combined, 48%, 27%, and 9.1% of the radioactive dose was excreted as unchanged brigatinib, N-desmethyl brigatinib (AP26123), and brigatinib cysteine conjugate, respectively. Unchanged brigatinib was the major circulating radioactive component (92%) along with AP26123 (3.5%), the primary metabolite also observed in vitro. In patients, at steady state, the plasma AUC of AP26123 was < 10% of brigatinib exposure. In in vitro kinase and cellular assays, the metabolite, AP26123, inhibited ALK with approximately 3-fold lower potency than brigatinib.
Elimination: In patients given brigatinib 180 mg once daily, the geometric mean apparent oral clearance (CL/F) of brigatinib at steady state was 8.9 L/h and the median plasma elimination half-life was 24 h.
The primary route of excretion of brigatinib is in faeces. In six healthy male subjects given a single 180 mg oral dose of [
14C]brigatinib, 65% of the administered dose was recovered in faeces and 25% of the administered dose was recovered in urine. Unchanged brigatinib represented 41% and 86% of the total radioactivity in faeces and urine, respectively, the remainder being metabolites.
Special Populations: Hepatic impairment: The pharmacokinetics of brigatinib was characterised in healthy subjects with normal hepatic function (N = 9), and patients with mild hepatic impairment (Child-Pugh class A, N = 6), moderate hepatic impairment (Child-Pugh class B, N = 6), or severe hepatic impairment (Child-Pugh class C, N = 6). The pharmacokinetics of brigatinib was similar between healthy subjects with normal hepatic function and patients with mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment. Unbound AUC
0-INF was 37% higher in patients with severe hepatic impairment (Child-Pugh class C) as compared to healthy subjects with normal hepatic function (see Dosage & Administration).
Renal impairment: The pharmacokinetics of brigatinib is similar in patients with normal renal function and in patients with mild or moderate renal impairment (eGFR ≥ 30 mL/min) based on the results of population pharmacokinetic analyses. In a pharmacokinetic study, unbound AUC
0-INF was 94% higher in patients with severe renal impairment (eGFR < 30 mL/min, N = 6) as compared to patients with normal renal function (eGFR ≥ 90 mL/min, N = 8) (see Dosage & Administration).
Race and gender: Population pharmacokinetic analyses showed that race and gender had no impact on the pharmacokinetics of brigatinib.
Age, body weight, and albumin concentrations: The population pharmacokinetic analyses showed that body weight, age, and albumin concentration had no clinically relevant impact on the pharmacokinetics of brigatinib.
Toxicology: Preclinical safety data: Safety pharmacology studies with brigatinib identified potential for pulmonary effects (altered respiration rate; 1-2 times the human C
max), cardiovascular effects (altered heart rate and blood pressure; at 0.5 times the human C
max), and renal effects (reduced renal function; at 1-2.5 times the human C
max), but did not indicate any potential for QT prolongation or neurofunctional effects.
Adverse reactions seen in animals at exposure levels similar to clinical exposure levels with possible relevance to clinical use were as follows: gastrointestinal system, bone marrow, eyes, testes, liver, kidney, bone, and heart. These effects were generally reversible during the non-dosing recovery period; however, effects in the eyes and testes were notable exceptions due to lack of recovery.
In repeated dose toxicity studies, lung changes (foamy alveolar macrophages) were noted in monkeys at ≥ 0.2 times the human AUC; however, these were minimal and similar to those reported as background findings in naive monkeys, and there was no clinical evidence of respiratory distress in these monkeys.
Carcinogenicity studies have not been performed with brigatinib.
Brigatinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) or the mammalian cell chromosomal aberration assays, but slightly increased the number of micronuclei in a rat bone marrow micronucleus test. The mechanism of micronucleus induction was abnormal chromosome segregation (aneugenicity) and not a clastogenic effect on chromosomes. This effect was observed at approximately five fold the human exposure at the 180 mg once daily dose.
Brigatinib may impair male fertility. Testicular toxicity was observed in repeat-dose animal studies. In rats, findings included lower weight of testes, seminal vesicles and prostate gland, and testicular tubular degeneration; these effects were not reversible during the recovery period. In monkeys, findings included reduced size of testes along with microscopic evidence of hypospermatogenesis; these effects were reversible during the recovery period. Overall, these effects on the male reproductive organs in rats and monkeys occurred at exposures ≥ 0.2-times the AUC observed in patients at the 180 mg once daily dose. No apparent adverse effects on female reproductive organs were observed in general toxicology studies in rats and monkeys.
In an embryo-foetal development study in which pregnant rats were administered daily doses of brigatinib during organogenesis; dose-related skeletal anomalies were observed at doses as low as approximately 0.7-times the human exposure by AUC at the 180 mg once daily dose. Findings included embryo-lethality, reduced foetal growth, and skeletal variations.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image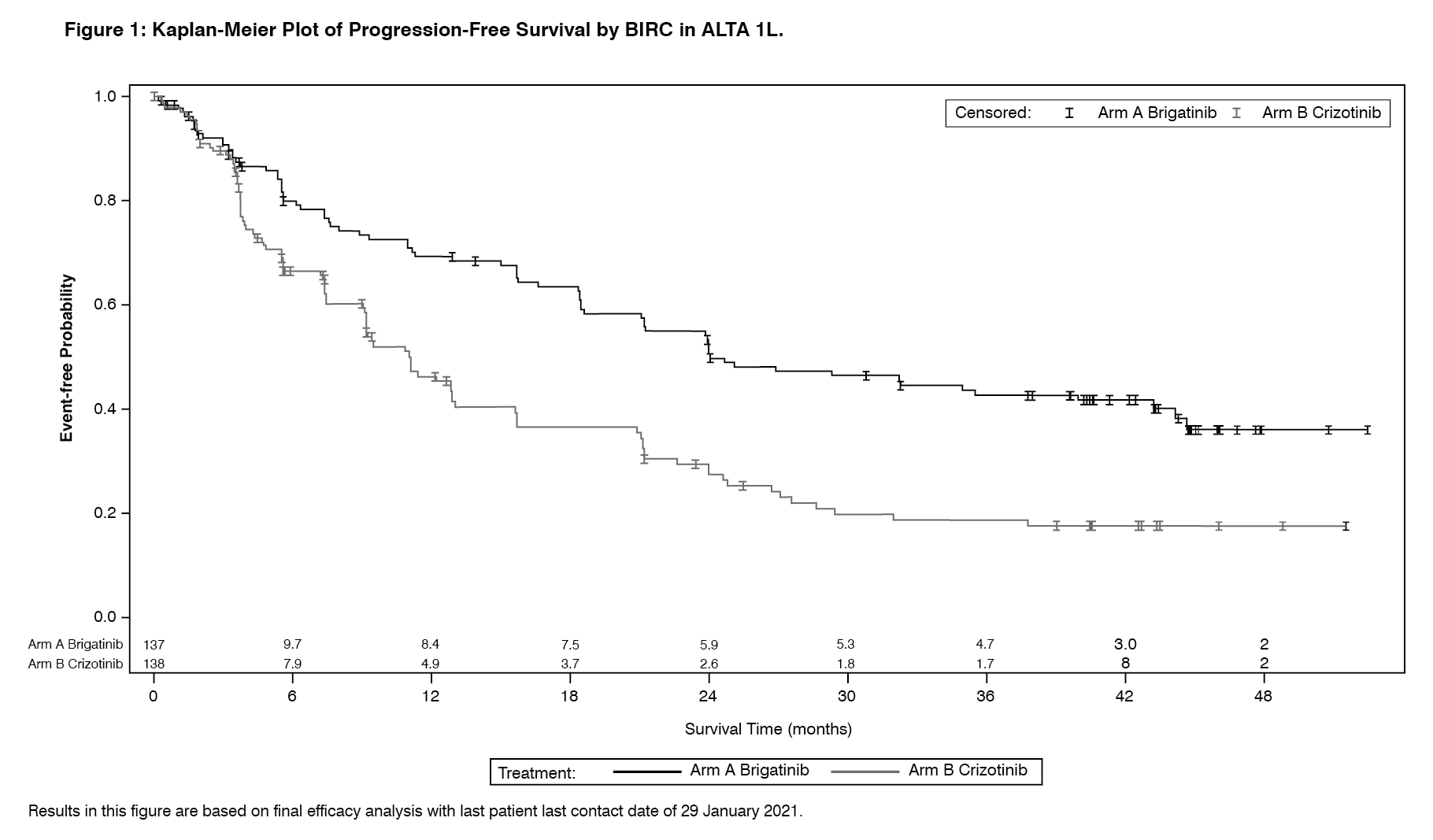 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image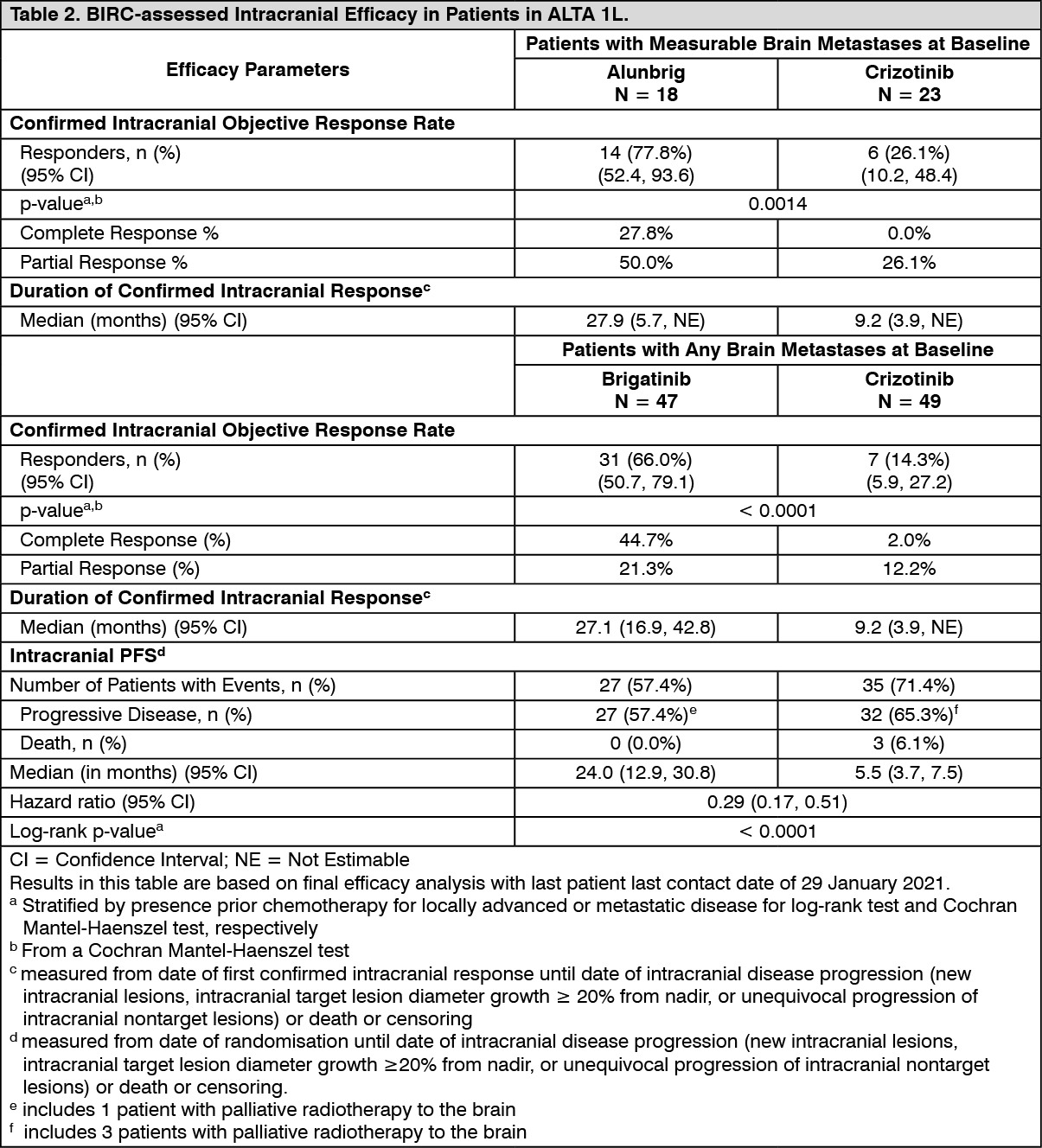 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image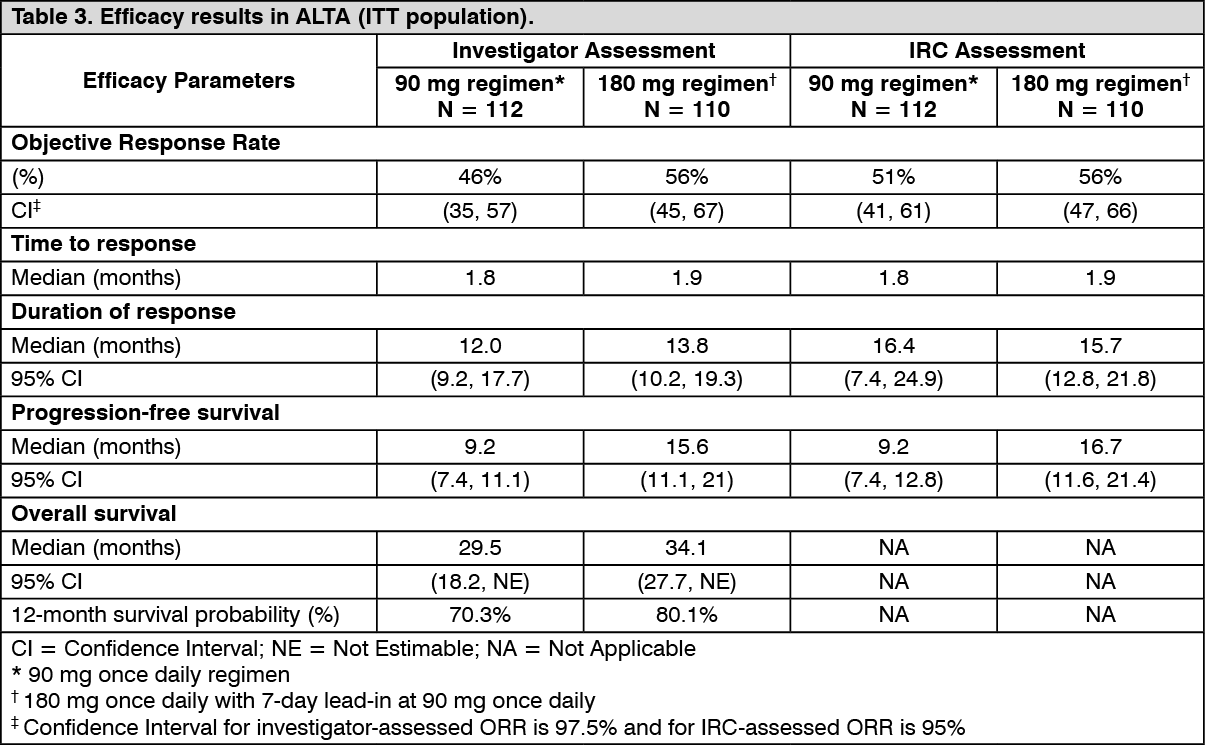 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image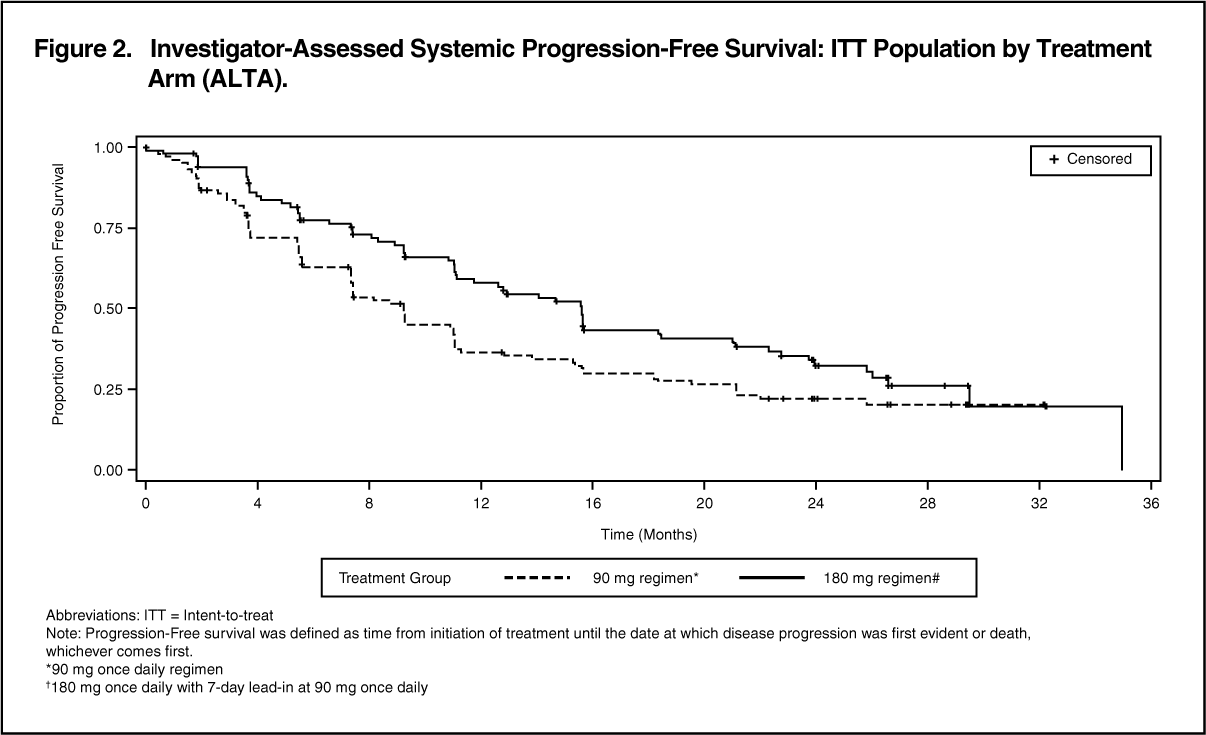 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image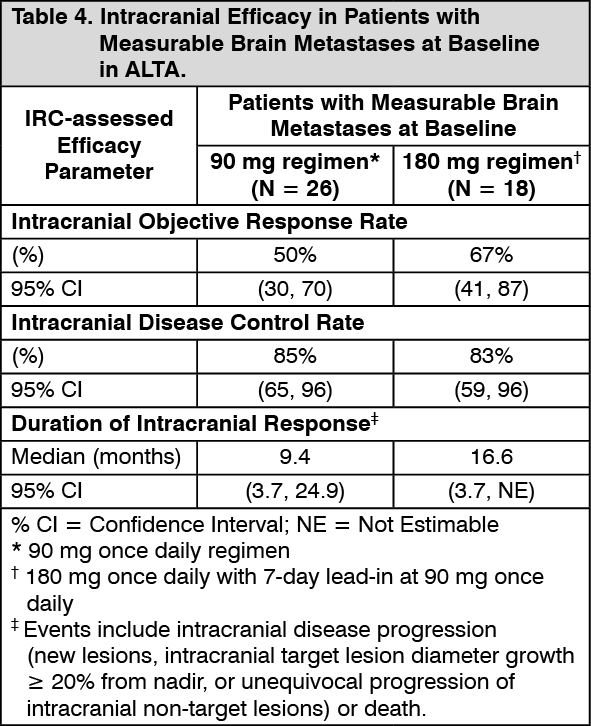 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image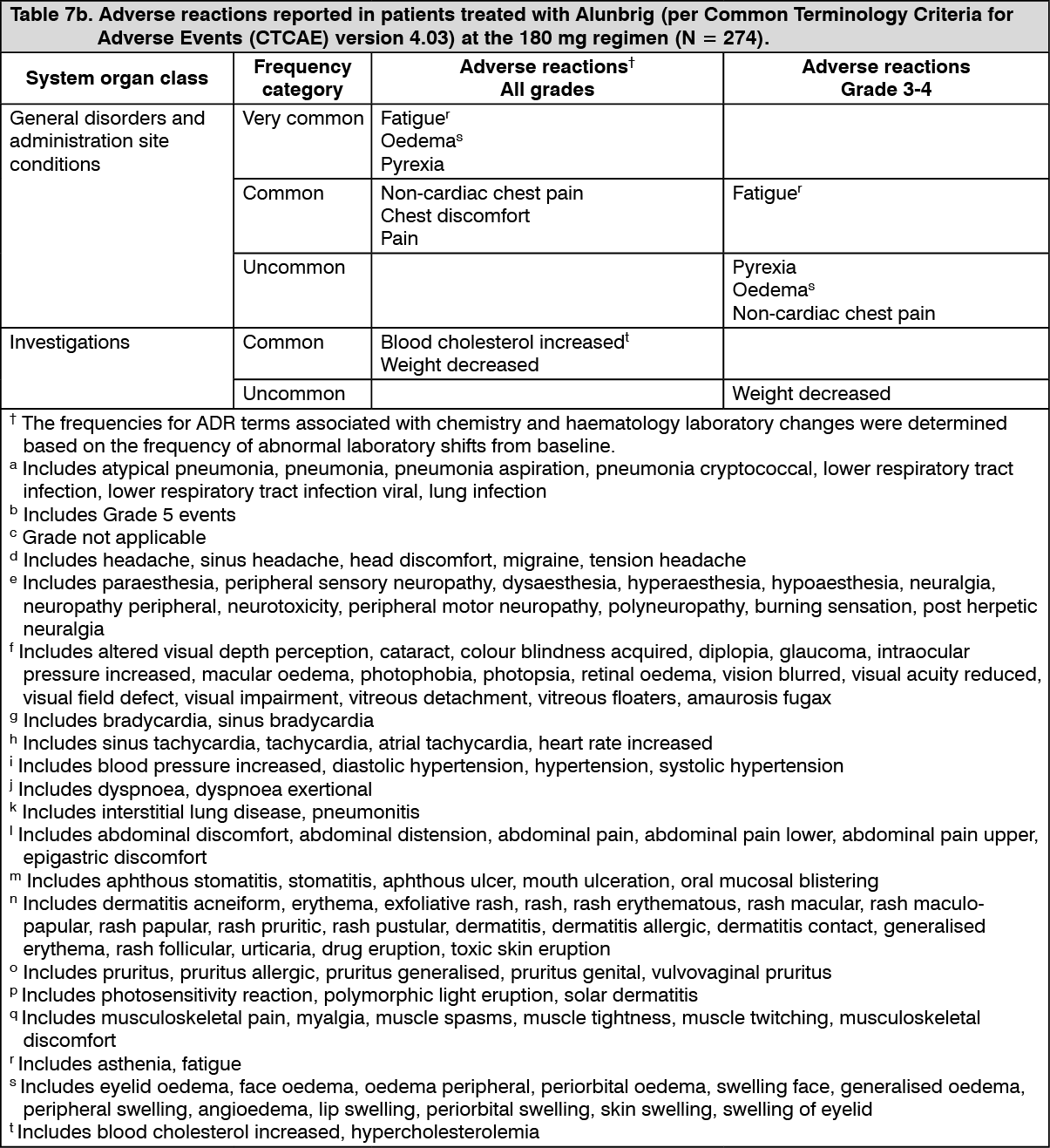 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out
